Multimodal analysis
Here we will use dual-omics SHARE-seq dataset, more specicially the dataset from figure4 in SHARE-seq study, “multiome_ma2020_fig4” as an example to illustrate how SIMBA performs multimodal analysis
[1]:
import os
import simba as si
si.__version__
[1]:
'1.0'
[2]:
workdir = 'result_multiome_shareseq'
si.settings.set_workdir(workdir)
Saving results in: result_multiome_shareseq
[3]:
si.settings.set_figure_params(dpi=80,
style='white',
fig_size=[5,5],
rc={'image.cmap': 'viridis'})
[4]:
# to make plots prettier
from IPython.display import set_matplotlib_formats
set_matplotlib_formats('retina')
[ ]:
load example data
[5]:
dict_adata = si.datasets.multiome_ma2020_fig4()
multiome_ma2020_fig4_rna.h5ad: 0.00B [00:00, ?B/s]
Downloading data ...
multiome_ma2020_fig4_rna.h5ad: 43.7MB [00:27, 1.60MB/s]
multiome_ma2020_fig4_atac.h5ad: 0.00B [00:00, ?B/s]
Downloaded to result_multiome_shareseq/data.
Downloading data ...
multiome_ma2020_fig4_atac.h5ad: 287MB [02:39, 1.79MB/s]
Downloaded to result_multiome_shareseq/data.
[6]:
dict_adata
[6]:
{'rna': AnnData object with n_obs × n_vars = 6436 × 20331
obs: 'celltype',
'atac': AnnData object with n_obs × n_vars = 6436 × 344592
obs: 'depth', 'FRIP', 'TSSportion', 'EnhancerPortion', 'atac.barcode', 'rna.barcode', 'atac.umap1', 'atac.umap2', 'celltype', 'rna.umap1', 'rna.umap2'
var: 'chr', 'start', 'end', 'width', 'strand', 'TSSidx', 'Enhanceridx', 'Gene', 'peaks'}
[7]:
adata_CP = dict_adata['atac']
adata_CG = dict_adata['rna']
[8]:
adata_CP.obs.head()
[8]:
| depth | FRIP | TSSportion | EnhancerPortion | atac.barcode | rna.barcode | atac.umap1 | atac.umap2 | celltype | rna.umap1 | rna.umap2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Trial60.skin.R1.01.R2.06.R3.83.P1.55 | 8135 | 0.702274 | 0.311920 | 0.265185 | Trial60.skin.R1.01.R2.06.R3.83.P1.07 | Trial60.skin.R1.01.R2.06.R3.83.P1.55 | 1.287024 | -9.679595 | TAC-1 | 0.801948 | -8.777440 |
| Trial60.skin.R1.01.R2.11.R3.86.P1.56 | 12614 | 0.709846 | 0.356489 | 0.248046 | Trial60.skin.R1.01.R2.11.R3.86.P1.08 | Trial60.skin.R1.01.R2.11.R3.86.P1.56 | 1.679156 | -8.890386 | TAC-1 | 0.576971 | -8.106592 |
| Trial60.skin.R1.01.R2.15.R3.09.P1.55 | 11424 | 0.799545 | 0.334465 | 0.261222 | Trial60.skin.R1.01.R2.15.R3.09.P1.07 | Trial60.skin.R1.01.R2.15.R3.09.P1.55 | 3.105708 | -11.200886 | TAC-1 | -0.170911 | -7.537184 |
| Trial60.skin.R1.01.R2.17.R3.81.P1.53 | 6588 | 0.802368 | 0.385736 | 0.234771 | Trial60.skin.R1.01.R2.17.R3.81.P1.05 | Trial60.skin.R1.01.R2.17.R3.81.P1.53 | 1.942233 | -9.400400 | TAC-1 | 0.778544 | -8.332716 |
| Trial60.skin.R1.01.R2.18.R3.95.P1.54 | 7796 | 0.733710 | 0.321154 | 0.259790 | Trial60.skin.R1.01.R2.18.R3.95.P1.06 | Trial60.skin.R1.01.R2.18.R3.95.P1.54 | 0.205076 | -10.888144 | TAC-1 | 1.867226 | -6.767437 |
[9]:
adata_CG.obs.head()
[9]:
| celltype | |
|---|---|
| Trial60.skin.R1.01.R2.06.R3.83.P1.55 | TAC-1 |
| Trial60.skin.R1.01.R2.11.R3.86.P1.56 | TAC-1 |
| Trial60.skin.R1.01.R2.15.R3.09.P1.55 | TAC-1 |
| Trial60.skin.R1.01.R2.17.R3.81.P1.53 | TAC-1 |
| Trial60.skin.R1.01.R2.18.R3.95.P1.54 | TAC-1 |
[ ]:
ATAC-seq part
preprocessing
[12]:
si.pp.filter_peaks(adata_CP,min_n_cells=3)
Before filtering:
6436 cells, 344592 peaks
Filter peaks based on min_n_cells
/data/pinello/SHARED_SOFTWARE/anaconda_latest/envs/hc_simba/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
After filtering out low-expressed peaks:
6436 cells, 332987 peaks
[13]:
si.pp.cal_qc_atac(adata_CP)
[14]:
si.pl.violin(adata_CP,list_obs=['n_counts','n_peaks','pct_peaks'], list_var=['n_cells'])
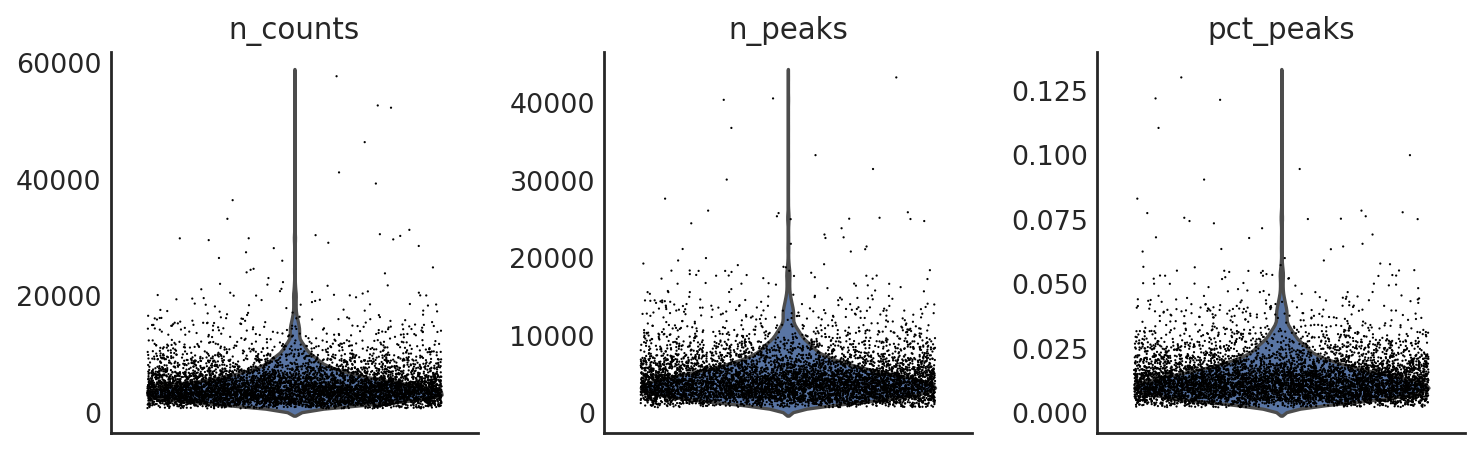
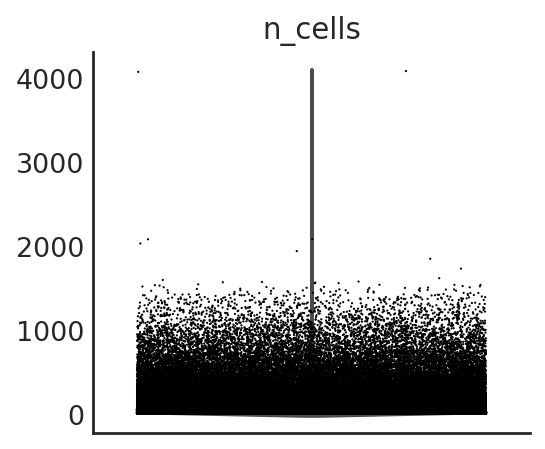
[15]:
si.pl.hist(adata_CP,list_obs=['n_counts','n_peaks','pct_peaks'], log=True, list_var=['n_cells'])
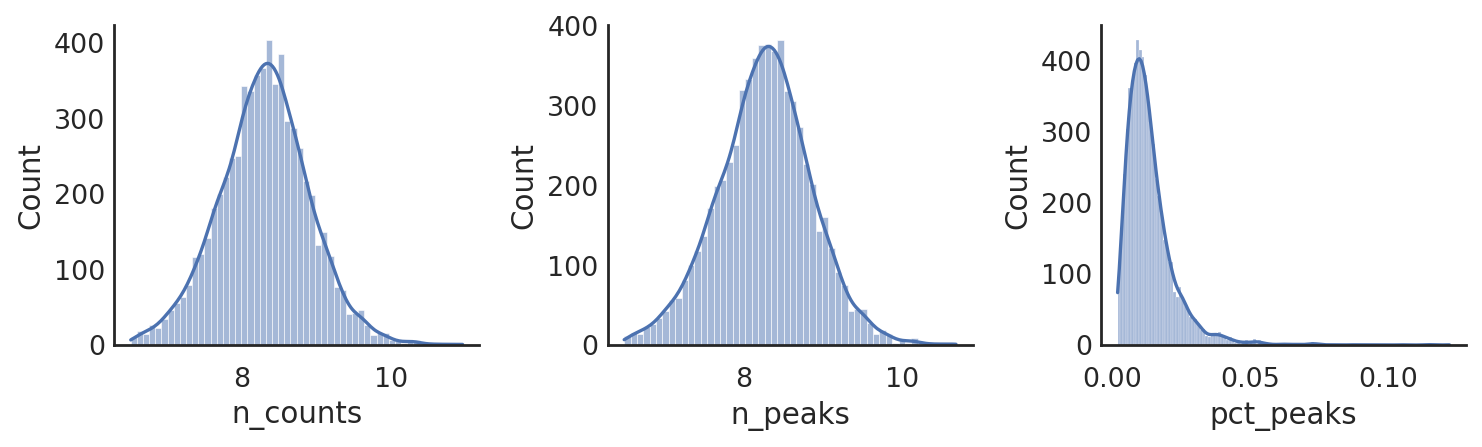
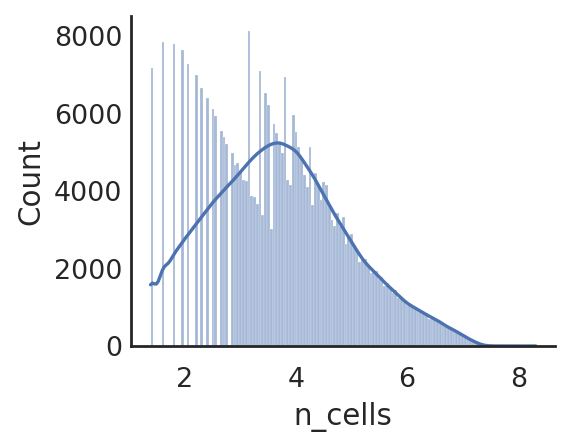
[ ]:
si.pp.pca(adata_CP, n_components=40)
si.pl.pca_variance_ratio(adata_CP)
si.pp.select_pcs_features(adata_CP)
[ ]:
select DNA sequences (Optional)
DNA sequences such as TF motifs or k-mers can be also encoded into the graph if needed.
To scan peaks for kmers and motifs, first write the peaks in a bed file.
[16]:
si.write_bed(adata_CP, use_top_pcs=False)
"peaks.bed" was written to "result_multiome_shareseq".
Then run the provided R script “scan_for_kmers_motifs.R” under the result directory (‘mm10.fa’ can be downloaded here)
[17]:
os.chdir(workdir)
[18]:
#! wget https://hgdownload.soe.ucsc.edu/goldenPath/mm10/bigZips/mm10.fa.gz
#! gunzip mm10.fa.gz
! wget https://raw.githubusercontent.com/pinellolab/simba/master/R_scripts/scan_for_kmers_motifs.R
--2021-11-09 16:48:26-- https://raw.githubusercontent.com/pinellolab/simba/master/R_scripts/scan_for_kmers_motifs.R
Resolving raw.githubusercontent.com (raw.githubusercontent.com)... 185.199.108.133, 185.199.109.133, 185.199.110.133, ...
Connecting to raw.githubusercontent.com (raw.githubusercontent.com)|185.199.108.133|:443... connected.
HTTP request sent, awaiting response... 200 OK
Length: 5767 (5.6K) [text/plain]
Saving to: ‘scan_for_kmers_motifs.R.1’
100%[======================================>] 5,767 --.-K/s in 0s
2021-11-09 16:48:26 (12.1 MB/s) - ‘scan_for_kmers_motifs.R.1’ saved [5767/5767]
Install the required packages in your enviroment with the following command:
$ conda install r-essentials r-optparse bioconductor-jaspar2020 bioconductor-biostrings bioconductor-tfbstools bioconductor-motifmatchr bioconductor-summarizedexperiment r-doparallel bioconductor-rhdf5 bioconductor-hdf5array
[19]:
%time
! Rscript scan_for_kmers_motifs.R -i peaks.bed -g mm10.fa -s 'Mus musculus'
CPU times: user 7 µs, sys: 1 µs, total: 8 µs
Wall time: 14.8 µs
[1] "Converting .bed to .fasta ..."
[1] "Scanning for kmers ..."
[1] "Scanning for TF motifs ..."
[1] "Saving kmer matrix ..."
[1] "Saving motif matrix ..."
[1] "Finished."
[20]:
os.chdir("../")
[ ]:
[21]:
adata_PK = si.read_hdf(os.path.join(workdir,'output_kmers_motifs/freq_kmer.h5'),'mat')
adata_PM = si.read_hdf(os.path.join(workdir,'output_kmers_motifs/freq_motif.h5'),'mat')
# convert byte string to string
adata_PK.obs.index = [x.decode('utf-8') for x in adata_PK.obs.index]
adata_PK.var.index = [x.decode('utf-8') for x in adata_PK.var.index]
adata_PM.obs.index = [x.decode('utf-8') for x in adata_PM.obs.index]
adata_PM.var.index = [x.decode('utf-8') for x in adata_PM.var.index]
[22]:
adata_PK
[22]:
AnnData object with n_obs × n_vars = 332987 × 4096
[23]:
adata_PM
[23]:
AnnData object with n_obs × n_vars = 332987 × 884
[24]:
si.pp.binarize(adata_PK)
si.pp.binarize(adata_PM)
[ ]:
select kmers and motifs (optional)
si.pp.pca(adata_PK, n_components=30)
si.pp.pca(adata_PM, n_components=30)
si.pp.select_pcs_features(adata_PK, min_elbow=adata_PK.shape[1]/5, S=5)
si.pp.select_pcs_features(adata_PM, min_elbow=adata_PM.shape[1]/5, S=5)
[ ]:
RNA-seq part
preprocessing
[25]:
si.pp.filter_genes(adata_CG,min_n_cells=3)
Before filtering:
6436 cells, 20331 genes
Filter genes based on min_n_cells
After filtering out low-expressed genes:
6436 cells, 17399 genes
/data/pinello/SHARED_SOFTWARE/anaconda_latest/envs/hc_simba/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
[26]:
si.pp.cal_qc_rna(adata_CG)
[27]:
si.pp.normalize(adata_CG,method='lib_size')
[28]:
si.pp.log_transform(adata_CG)
[ ]:
Optionally, variable gene selection step can be also performed.
si.pp.select_variable_genes(adata_CG)
si.pl.variable_genes(adata_CG,show_texts=True)
This will speed up the training procedure as only variable genes are encoded into the graph. But we won’t obtain the embeddings of non-variable genes.
[ ]:
discretize RNA expression
[29]:
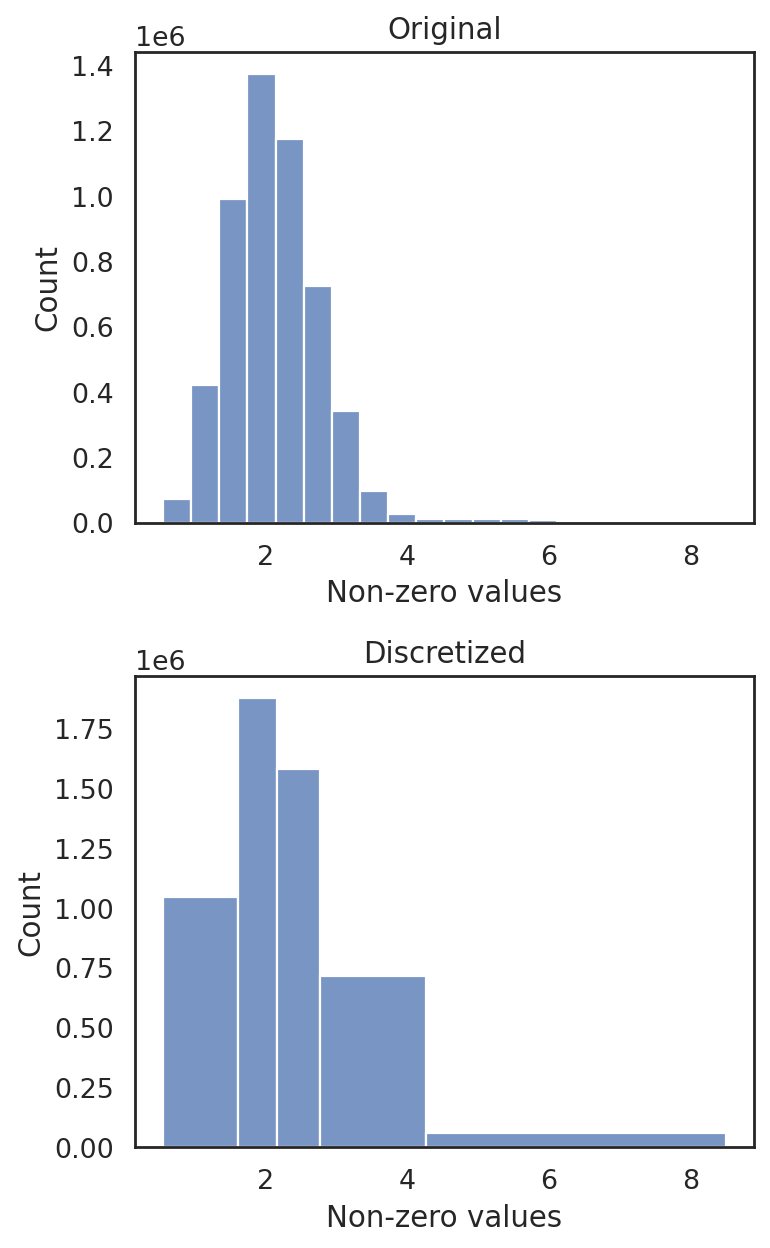
si.tl.discretize(adata_CG,n_bins=5)
[30]:
si.pl.discretize(adata_CG,kde=False)
[0.54990256 1.604449 2.161982 2.770135 4.260093 8.499525 ]
[ ]:
generate graph
[31]:
si.tl.gen_graph(list_CP=[adata_CP],
list_CG=[adata_CG],
list_PK=[adata_PK],
list_PM=[adata_PM],
copy=False,
use_highly_variable=False,
use_top_pcs=False,
dirname='graph0')
relation0: source: C, destination: P
#edges: 29715636
relation1: source: P, destination: M
#edges: 11851328
relation2: source: P, destination: K
#edges: 90598213
relation3: source: C, destination: G
#edges: 1046833
relation4: source: C, destination: G
#edges: 1877327
relation5: source: C, destination: G
#edges: 1580227
relation6: source: C, destination: G
#edges: 717081
relation7: source: C, destination: G
#edges: 59974
Total number of edges: 137446619
Writing graph file "pbg_graph.txt" to "result_multiome_shareseq/pbg/graph0" ...
Finished.
[ ]:
PBG training
Before PBG training, let’s take a look at the parameters:
[32]:
si.settings.pbg_params
[32]:
{'entity_path': 'result_multiome_shareseq/pbg/graph0/input/entity',
'edge_paths': ['result_multiome_shareseq/pbg/graph0/input/edge'],
'checkpoint_path': '',
'entities': {'C': {'num_partitions': 1},
'G': {'num_partitions': 1},
'P': {'num_partitions': 1},
'K': {'num_partitions': 1},
'M': {'num_partitions': 1}},
'relations': [{'name': 'r0',
'lhs': 'C',
'rhs': 'P',
'operator': 'none',
'weight': 1.0},
{'name': 'r1', 'lhs': 'P', 'rhs': 'M', 'operator': 'none', 'weight': 0.2},
{'name': 'r2', 'lhs': 'P', 'rhs': 'K', 'operator': 'none', 'weight': 0.02},
{'name': 'r3', 'lhs': 'C', 'rhs': 'G', 'operator': 'none', 'weight': 1.0},
{'name': 'r4', 'lhs': 'C', 'rhs': 'G', 'operator': 'none', 'weight': 2.0},
{'name': 'r5', 'lhs': 'C', 'rhs': 'G', 'operator': 'none', 'weight': 3.0},
{'name': 'r6', 'lhs': 'C', 'rhs': 'G', 'operator': 'none', 'weight': 4.0},
{'name': 'r7', 'lhs': 'C', 'rhs': 'G', 'operator': 'none', 'weight': 5.0}],
'dynamic_relations': False,
'dimension': 50,
'global_emb': False,
'comparator': 'dot',
'num_epochs': 10,
'workers': 4,
'num_batch_negs': 50,
'num_uniform_negs': 50,
'loss_fn': 'softmax',
'lr': 0.1,
'early_stopping': False,
'regularization_coef': 0.0,
'wd': 0.0,
'wd_interval': 50,
'eval_fraction': 0.05,
'eval_num_batch_negs': 50,
'eval_num_uniform_negs': 50,
'checkpoint_preservation_interval': None}
[ ]:
If no parameters need to be adjusted, the training can be simply done with:
si.tl.pbg_train(auto_wd=True, save_wd=True, output='model')
[ ]:
Here we show how to adjust training-related parameters if needed. In general, weight decay wd is the only parameter that might need to be adjusted based on the following pbg metric plots. However, in almost all the cases, the automatically decided wd (enabling it by setting auto_wd=True) works well.
E.g. we want to change the number of cpus workers:
[33]:
# modify parameters
dict_config = si.settings.pbg_params.copy()
# dict_config['wd'] = 0.000172
dict_config['workers'] = 12
## start training
si.tl.pbg_train(pbg_params = dict_config, auto_wd=True, save_wd=True, output='model')
Auto-estimated weight decay is 0.000172
`.settings.pbg_params['wd']` has been updated to 0.000172
Converting input data ...
[2021-06-25 16:19:24.210303] Found some files that indicate that the input data has already been preprocessed, not doing it again.
[2021-06-25 16:19:24.210611] These files are in: result_multiome_shareseq/pbg/graph0/input/entity, result_multiome_shareseq/pbg/graph0/input/edge
Starting training ...
Finished
[ ]:
If
wdis specified by users instead of being automatically decided, then make sure to update it in simba setting:
si.settings.pbg_params = dict_config.copy()
[ ]:
The trained result can be loaded in with the following steps:
By default, it’s using the current training result stored in .setting.pbg_params
# load in graph ('graph0') info
si.load_graph_stats()
# load in model info for ('graph0')
si.load_pbg_config()
Users can also specify different pathss
# load in graph ('graph0') info
si.load_graph_stats(path='./result_multiome_shareseq/pbg/graph0/')
# load in model info for ('graph0')
si.load_pbg_config(path='./result_multiome_shareseq/pbg/graph0/model/')
[ ]:
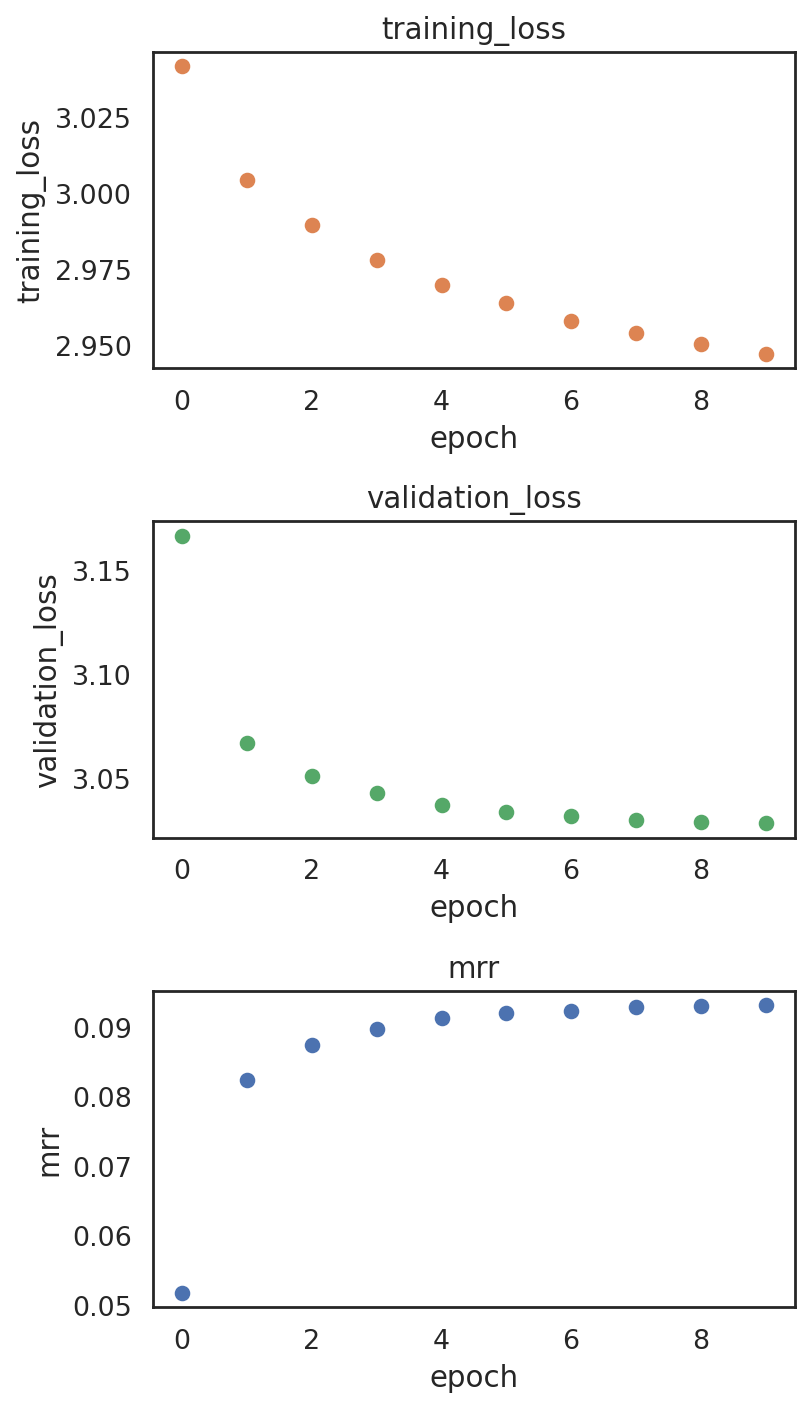
Plotting training metrics to make sure the model is not overfitting
[34]:
si.pl.pbg_metrics(fig_ncol=1)
[ ]:
post-training analysis
[35]:
palette_celltype={'TAC-1':'#F8D856', 'TAC-2':'#F1B044', 'IRS':'#C37777',
'Medulla':'#897a74','Hair Shaft-cuticle.cortex':"#d6a780"}
[36]:
dict_adata = si.read_embedding()
[37]:
dict_adata
[37]:
{'C': AnnData object with n_obs × n_vars = 6436 × 50,
'G': AnnData object with n_obs × n_vars = 17399 × 50,
'K': AnnData object with n_obs × n_vars = 4096 × 50,
'M': AnnData object with n_obs × n_vars = 884 × 50,
'P': AnnData object with n_obs × n_vars = 332987 × 50}
[39]:
adata_C = dict_adata['C'] # embeddings for cells
adata_G = dict_adata['G'] # embeddings for genes
adata_P = dict_adata['P'] # embeddings for peaks
adata_K = dict_adata['K'] # embeddings for kmers
adata_M = dict_adata['M'] # embeddings for motifs
#### to distinguish TF motif names from gene names in this case
adata_M.obs.index = 'M_'+adata_M.obs.index
[40]:
adata_C
[40]:
AnnData object with n_obs × n_vars = 6436 × 50
[41]:
adata_G
[41]:
AnnData object with n_obs × n_vars = 17399 × 50
[42]:
adata_P
[42]:
AnnData object with n_obs × n_vars = 332987 × 50
[43]:
adata_K
[43]:
AnnData object with n_obs × n_vars = 4096 × 50
[44]:
adata_M
[44]:
AnnData object with n_obs × n_vars = 884 × 50
[ ]:
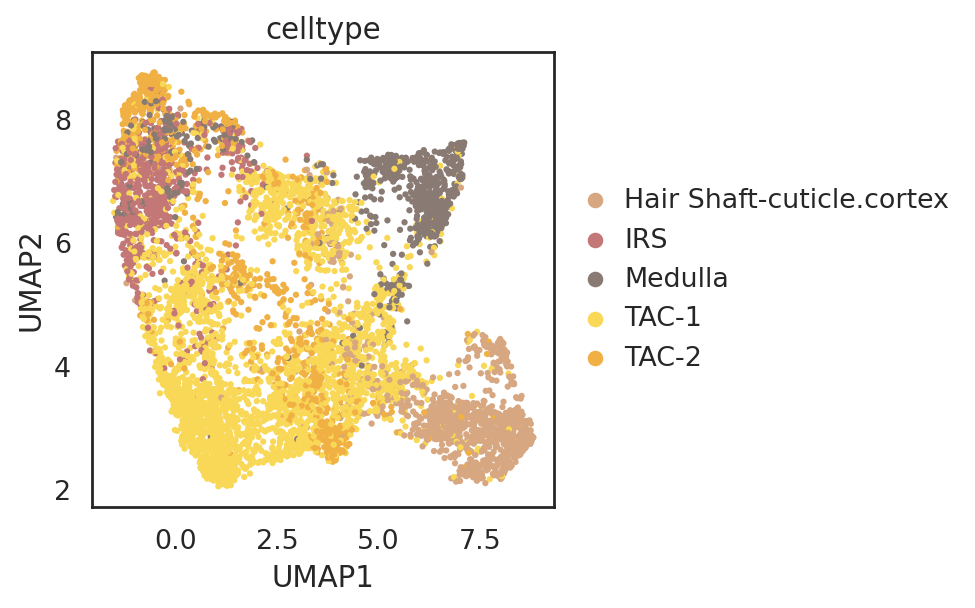
visualize embeddings of cells
[45]:
## Add annotation of celltypes (optional)
adata_C.obs['celltype'] = adata_CG[adata_C.obs_names,:].obs['celltype'].copy()
adata_C
/data/pinello/SHARED_SOFTWARE/anaconda_latest/envs/hc_simba/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
[45]:
AnnData object with n_obs × n_vars = 6436 × 50
obs: 'celltype'
[46]:
si.tl.umap(adata_C,n_neighbors=15,n_components=2)
[46]:
si.pl.umap(adata_C,
color=['celltype'],dict_palette={'celltype': palette_celltype},
fig_size=(6,4),
drawing_order='random')
[ ]:
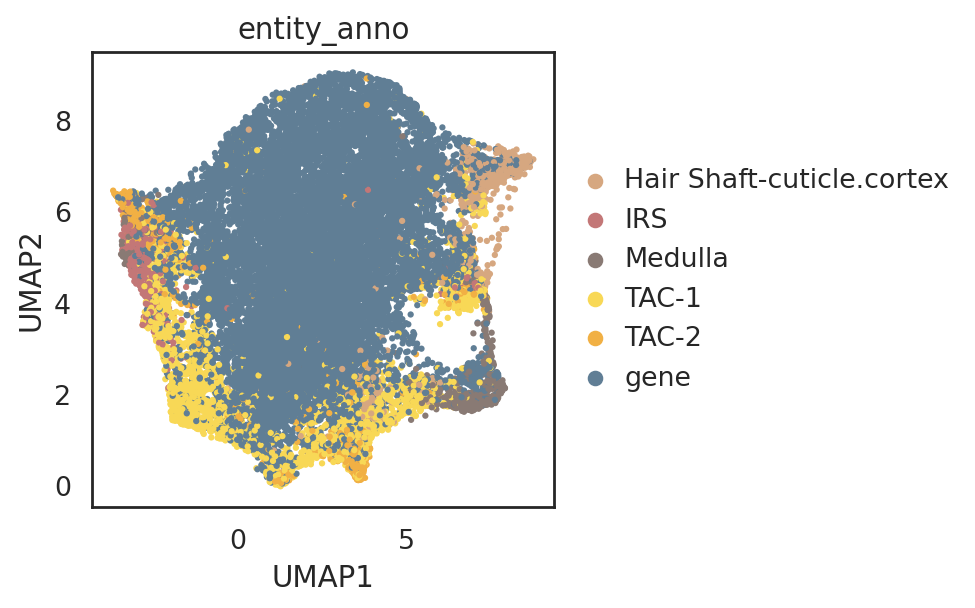
visualize embeddings of cells and genes
[47]:
adata_all_CG = si.tl.embed(adata_ref=adata_C,
list_adata_query=[adata_G])
Performing softmax transformation for query data 0;
[48]:
## add annotations of all entities
adata_all_CG.obs['entity_anno'] = ""
adata_all_CG.obs.loc[adata_C.obs_names, 'entity_anno'] = adata_all_CG.obs.loc[adata_C.obs_names, 'celltype'].tolist()
adata_all_CG.obs.loc[adata_G.obs_names, 'entity_anno'] = 'gene'
adata_all_CG.obs.head()
[48]:
| celltype | id_dataset | entity_anno | |
|---|---|---|---|
| Trial60.skin.R1.02.R2.47.R3.02.P1.54 | TAC-2 | ref | TAC-2 |
| Trial60.skin.R1.51.R2.48.R3.30.P1.54 | IRS | ref | IRS |
| Trial60.skin.R1.64.R2.52.R3.11.P1.54 | Medulla | ref | Medulla |
| Trial60.skin.R1.74.R2.04.R3.13.P1.55 | TAC-1 | ref | TAC-1 |
| Trial60.skin.R1.94.R2.40.R3.61.P1.55 | Hair Shaft-cuticle.cortex | ref | Hair Shaft-cuticle.cortex |
[49]:
si.tl.umap(adata_all_CG,n_neighbors=15,n_components=2)
[50]:
palette_entity_anno = palette_celltype.copy()
palette_entity_anno['gene'] = "#607e95"
[51]:
si.pl.umap(adata_all_CG,
color=['entity_anno'],
dict_palette={'entity_anno': palette_entity_anno},
fig_size=(6,4),
drawing_order='random')
[52]:
#some marker genes from the original study
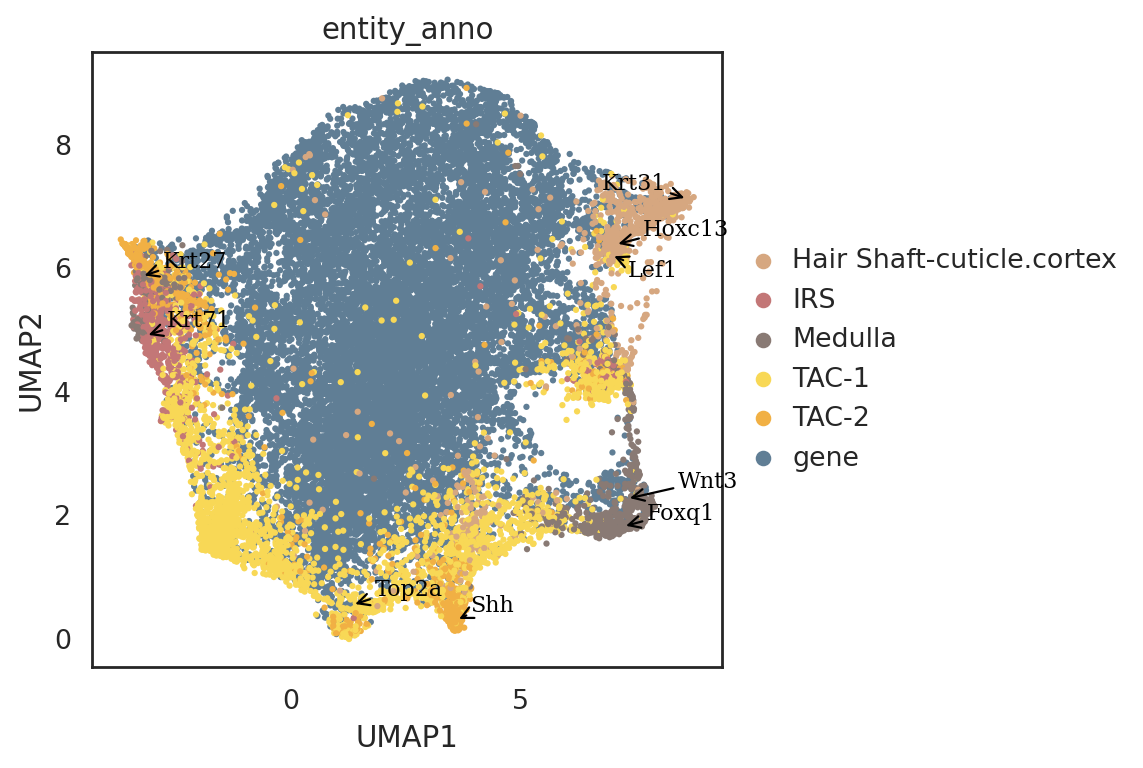
marker_genes = ['Wnt3','Top2a','Shh','Krt27','Foxq1', 'Krt31','Krt71', 'Lef1', 'Hoxc13']
[53]:
si.pl.umap(adata_all_CG[::-1,],
color=['entity_anno'],
dict_palette={'entity_anno': palette_entity_anno},
fig_size=(7,5),
texts=marker_genes,
text_expand=(2,1.5),
show_texts=True,
drawing_order='original')
/data/pinello/SHARED_SOFTWARE/anaconda_latest/envs/hc_simba/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
[ ]:
visualize embeddings of all entities including cells, genes, motifs, kmers, and peaks
[56]:
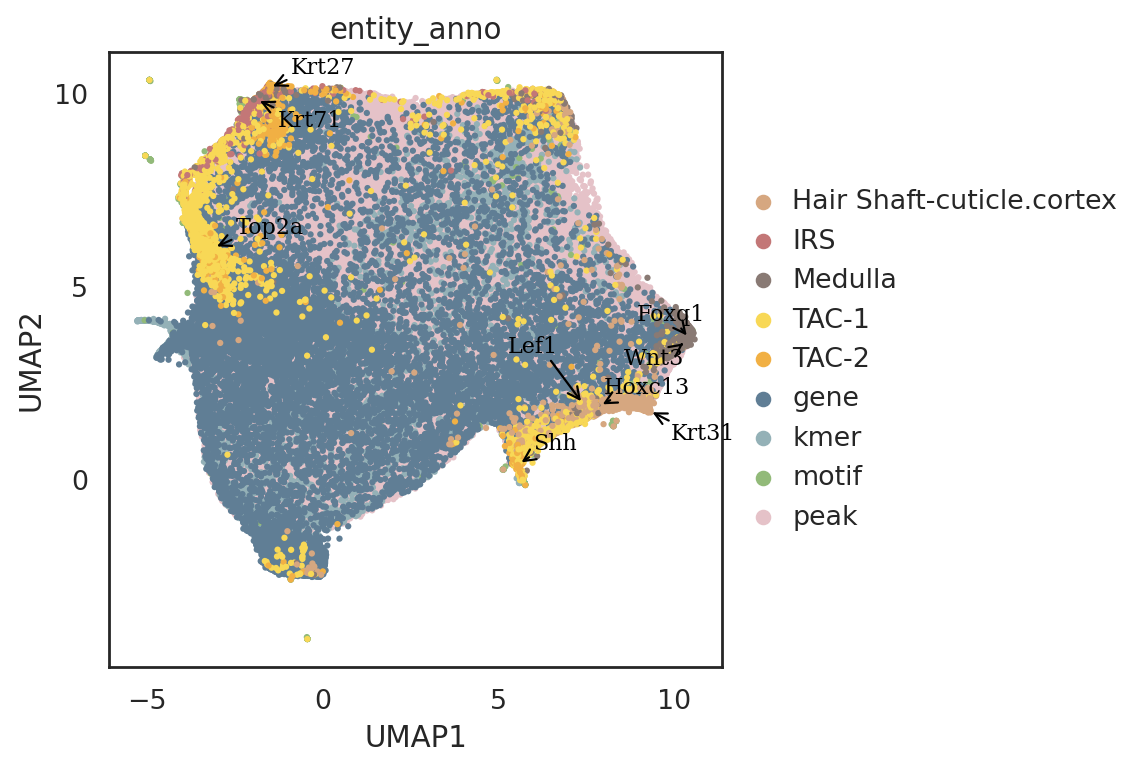
adata_all = si.tl.embed(adata_ref=adata_C,
list_adata_query=[adata_G, adata_M, adata_K, adata_P])
Performing softmax transformation for query data 0;
Performing softmax transformation for query data 1;
Performing softmax transformation for query data 2;
Performing softmax transformation for query data 3;
[57]:
## add annotations of all entities
adata_all.obs['entity_anno'] = ""
adata_all.obs.loc[adata_C.obs_names, 'entity_anno'] = adata_all.obs.loc[adata_C.obs_names, 'celltype'].tolist()
adata_all.obs.loc[adata_G.obs_names, 'entity_anno'] = 'gene'
adata_all.obs.loc[adata_P.obs_names, 'entity_anno'] = 'peak'
adata_all.obs.loc[adata_K.obs_names, 'entity_anno'] = 'kmer'
adata_all.obs.loc[adata_M.obs_names, 'entity_anno'] = 'motif'
adata_all.obs.head()
[57]:
| celltype | id_dataset | entity_anno | |
|---|---|---|---|
| Trial60.skin.R1.02.R2.47.R3.02.P1.54 | TAC-2 | ref | TAC-2 |
| Trial60.skin.R1.51.R2.48.R3.30.P1.54 | IRS | ref | IRS |
| Trial60.skin.R1.64.R2.52.R3.11.P1.54 | Medulla | ref | Medulla |
| Trial60.skin.R1.74.R2.04.R3.13.P1.55 | TAC-1 | ref | TAC-1 |
| Trial60.skin.R1.94.R2.40.R3.61.P1.55 | Hair Shaft-cuticle.cortex | ref | Hair Shaft-cuticle.cortex |
[61]:
si.tl.umap(adata_all,n_neighbors=50,n_components=2,n_jobs=20)
[62]:
palette_entity_anno = palette_celltype.copy()
palette_entity_anno['gene'] = "#607e95"
palette_entity_anno['kmer'] = "#94b1b7"
palette_entity_anno['motif'] = "#92ba79"
palette_entity_anno['peak'] = "#e5c2c8"
[63]:
si.pl.umap(adata_all[::-1,],
color=['entity_anno'],
dict_palette={'entity_anno': palette_entity_anno},
fig_size=(7,5),
drawing_order='original',
texts=marker_genes,
text_expand=(2,2),
show_texts=True)
/data/pinello/SHARED_SOFTWARE/anaconda_latest/envs/hc_simba/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
Trying to set attribute `.uns` of view, copying.
SIMBA metrics
[71]:
# genes
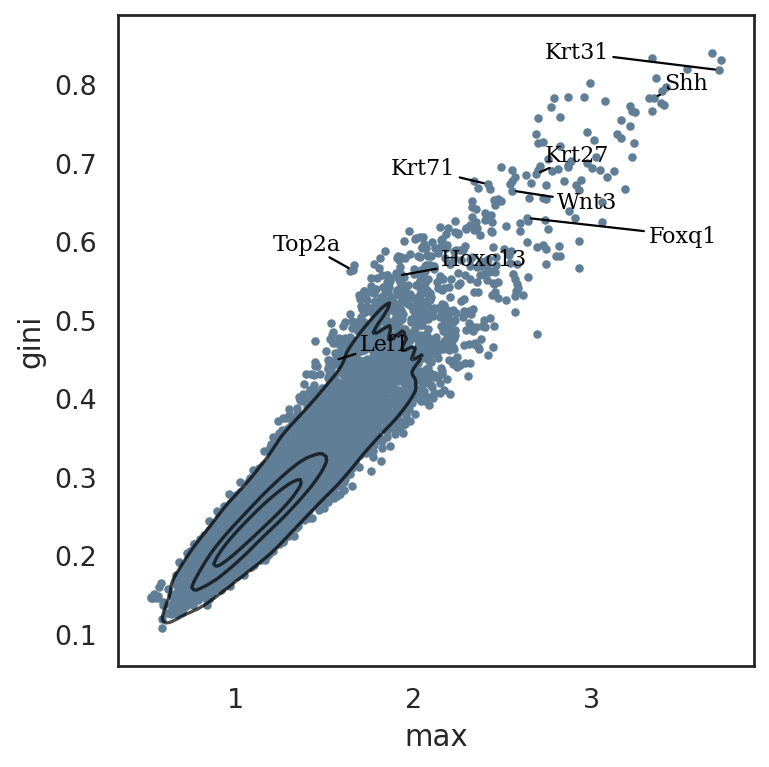
adata_cmp_CG = si.tl.compare_entities(adata_ref=adata_C,
adata_query=adata_G)
# motifs
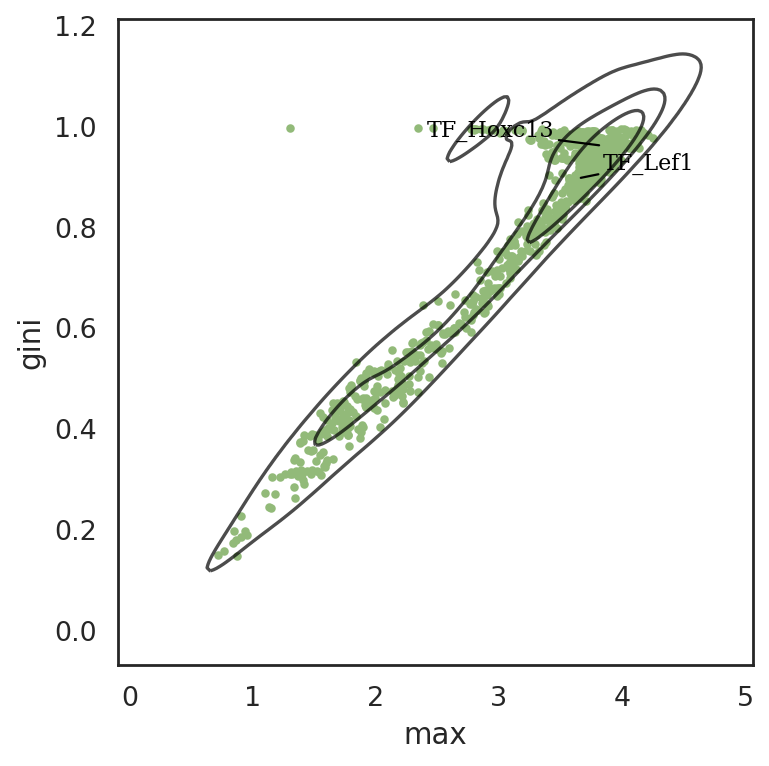
adata_cmp_CM = si.tl.compare_entities(adata_ref=adata_C,
adata_query=adata_M)
# peaks
adata_cmp_CP = si.tl.compare_entities(adata_ref=adata_C,
adata_query=adata_P)
[72]:
# rename TF motifs and peaks shorter names
import pandas as pd
adata_cmp_CM.var.index = \
pd.Series(adata_cmp_CM.var.index).replace(
to_replace=['M_ENSMUSG00000027985_LINE1723_Lef1_D', 'M_ENSMUSG00000001655_LINE1151_Hoxc13_D'],
value=['TF_Lef1', 'TF_Hoxc13'])
adata_cmp_CP.var.index = \
pd.Series(adata_cmp_CP.var.index).replace(
to_replace=['chr3_131018470_131018770', 'chr3_131104928_131105228', 'chr3_131177880_131178180', 'chr3_131212270_131212570',
'chr15_102832980_102833280', 'chr15_102855927_102856227'],
value=['Peak1(Lef1)', 'Peak2(Lef1)', 'Peak3(Lef1)', 'Peak4(Lef1)',
'Peak1(Hoxc13)', 'Peak2(Hoxc13)'])
adata_all.obs.index = \
pd.Series(adata_all.obs.index).replace(
to_replace=['M_ENSMUSG00000027985_LINE1723_Lef1_D', 'M_ENSMUSG00000001655_LINE1151_Hoxc13_D',
'chr3_131018470_131018770', 'chr3_131104928_131105228', 'chr3_131177880_131178180', 'chr3_131212270_131212570',
'chr15_102832980_102833280', 'chr15_102855927_102856227'],
value=['TF_Lef1', 'TF_Hoxc13',
'Peak1(Lef1)', 'Peak2(Lef1)', 'Peak3(Lef1)', 'Peak4(Lef1)',
'Peak1(Hoxc13)', 'Peak2(Hoxc13)'])
[73]:
si.pl.entity_metrics(adata_cmp_CG,
x='max',
y='gini',
show_texts=True,
show_cutoff=False,
show_contour=True,
c='#607e95',
texts=marker_genes,
text_expand=(2,1.5))
[74]:
si.pl.entity_metrics(adata_cmp_CM,
x='max',
y='gini',
show_texts=True,
show_cutoff=False,
show_contour=True,
c='#92ba79',
texts=['TF_Lef1','TF_Hoxc13'],
text_expand=(2,1.5))
[75]:
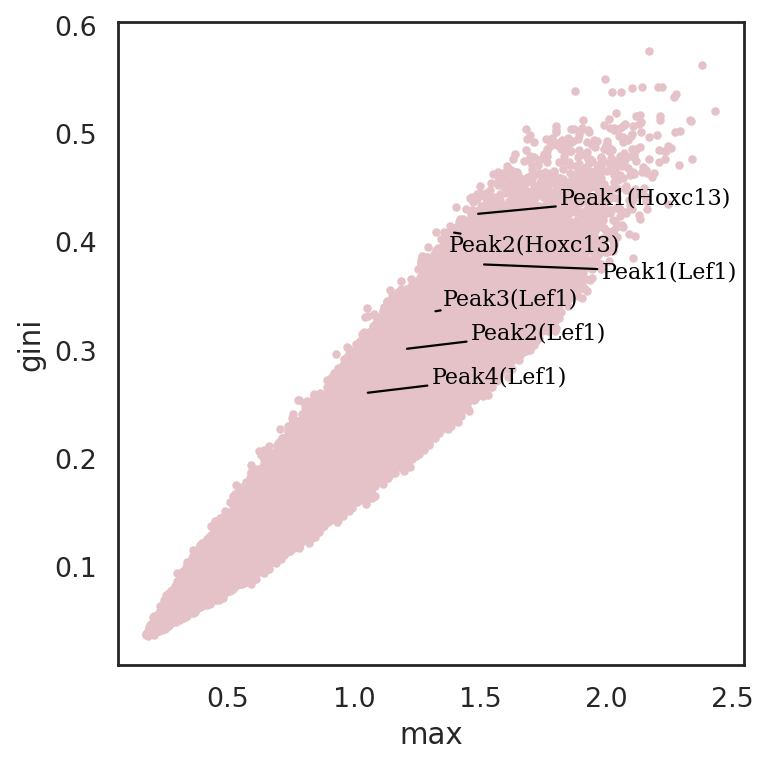
si.pl.entity_metrics(adata_cmp_CP,
x='max',
y='gini',
show_texts=True,
show_cutoff=False,
show_contour=False,
texts=['Peak1(Lef1)', 'Peak2(Lef1)', 'Peak3(Lef1)', 'Peak4(Lef1)',
'Peak1(Hoxc13)', 'Peak2(Hoxc13)'],
text_expand=(2,1.5),
c='#e5c2c8')
[ ]:
only visulize the embeddings of cells and cell-type specific features
[76]:
genes_selected = adata_cmp_CG.var[(adata_cmp_CG.var['max']>1.5) & (adata_cmp_CG.var['gini']>0.35)].index.tolist()
len(genes_selected)
[76]:
1960
[77]:
motifs_selected = adata_cmp_CM.var[(adata_cmp_CM.var['max']>3) & (adata_cmp_CM.var['gini']>0.7)].index.tolist()
len(motifs_selected)
[77]:
588
[78]:
# The union of top 1000 neighbor peaks of all marker genes and TF motifs
query_result = si.tl.query(adata_all,
obsm=None,
entity=marker_genes + ['TF_Lef1','TF_Hoxc13'],
k=1000,use_radius=False,
anno_filter='entity_anno',
filters=['peak'])
print(query_result.shape)
query_result.head()
peaks_selected = list(query_result.index.unique())
len(peaks_selected)
/data/pinello/SHARED_SOFTWARE/anaconda_latest/envs/hc_simba/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
(11000, 5)
[78]:
8619
[79]:
adata_all_selected = adata_all[adata_C.obs_names.to_list()
+ genes_selected
+ motifs_selected
+ peaks_selected, ].copy()
[82]:
si.tl.umap(adata_all_selected,n_neighbors=50,n_components=2)
[86]:
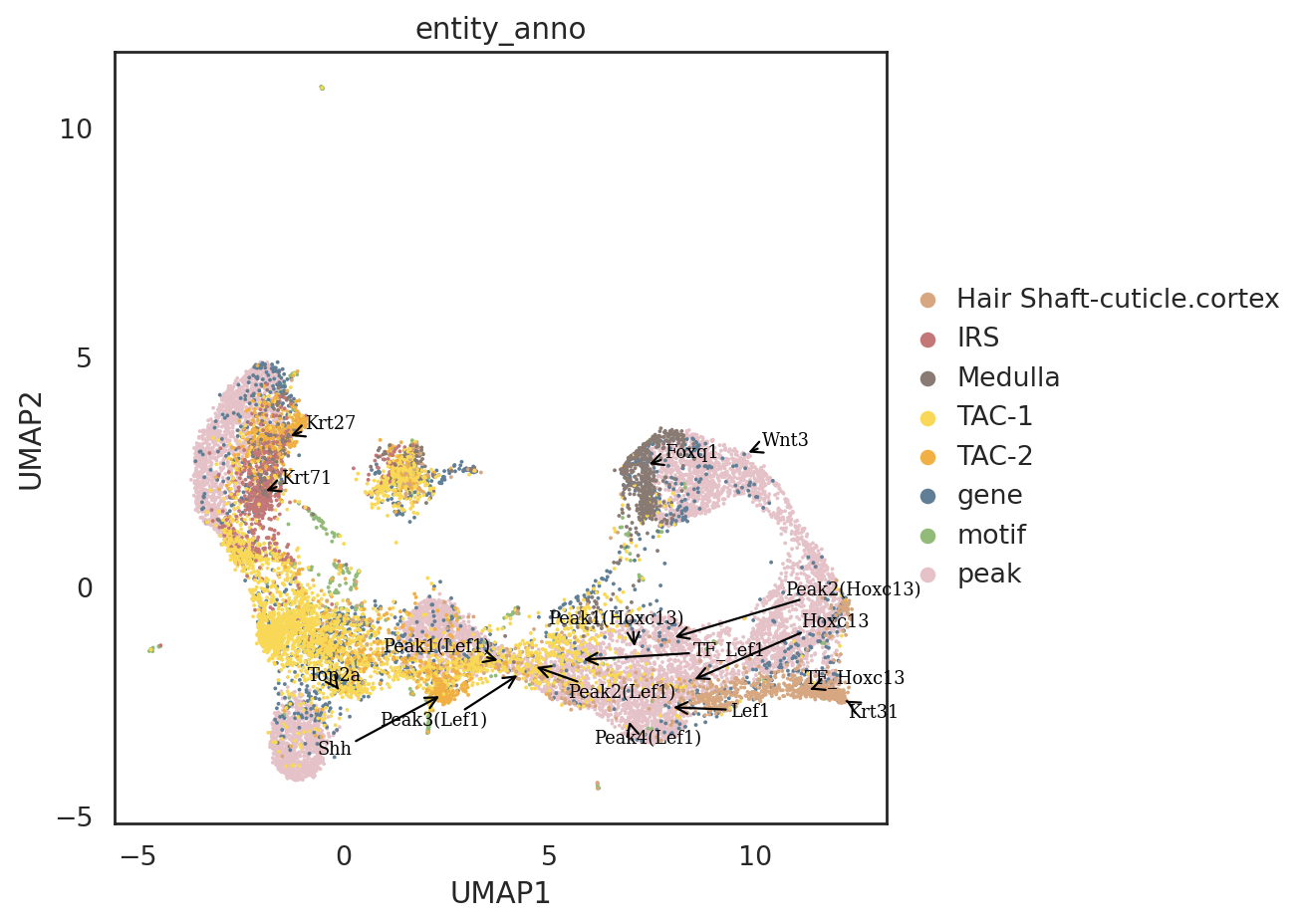
si.pl.umap(adata_all_selected[::-1,],
color=['entity_anno'],
dict_palette={'entity_anno': palette_entity_anno},
text_size=8,
fig_size=(8,6),
size=3,
drawing_order='original',
texts=marker_genes \
+ ['TF_Lef1','TF_Hoxc13'] \
+ ['Peak1(Lef1)', 'Peak2(Lef1)', 'Peak3(Lef1)', 'Peak4(Lef1)',
'Peak1(Hoxc13)', 'Peak2(Hoxc13)'],
text_expand=(2,1.5),
show_texts=True)
/data/pinello/SHARED_SOFTWARE/anaconda_latest/envs/hc_simba/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
Trying to set attribute `.uns` of view, copying.
[ ]:
save results
[87]:
# change shorter names back to the orignal names
adata_cmp_CM.var.index = \
pd.Series(adata_cmp_CM.var.index).replace(
to_replace =['TF_Lef1', 'TF_Hoxc13'],
value = ['M_ENSMUSG00000027985_LINE1723_Lef1_D', 'M_ENSMUSG00000001655_LINE1151_Hoxc13_D'])
adata_cmp_CP.var.index = \
pd.Series(adata_cmp_CP.var.index).replace(
to_replace = ['Peak1(Lef1)', 'Peak2(Lef1)', 'Peak3(Lef1)', 'Peak4(Lef1)', 'Peak1(Hoxc13)', 'Peak2(Hoxc13)'],
value = ['chr3_131018470_131018770', 'chr3_131104928_131105228', 'chr3_131177880_131178180', 'chr3_131212270_131212570',
'chr15_102832980_102833280', 'chr15_102855927_102856227'])
adata_all.obs.index = \
pd.Series(adata_all.obs.index).replace(
to_replace=['TF_Lef1', 'TF_Hoxc13',
'Peak1(Lef1)', 'Peak2(Lef1)', 'Peak3(Lef1)', 'Peak4(Lef1)',
'Peak1(Hoxc13)', 'Peak2(Hoxc13)'],
value=['M_ENSMUSG00000027985_LINE1723_Lef1_D', 'M_ENSMUSG00000001655_LINE1151_Hoxc13_D',
'chr3_131018470_131018770', 'chr3_131104928_131105228', 'chr3_131177880_131178180', 'chr3_131212270_131212570',
'chr15_102832980_102833280', 'chr15_102855927_102856227'])
adata_CG.write(os.path.join(workdir,'adata_CG.h5ad'))
adata_CP.write(os.path.join(workdir,'adata_CP.h5ad'))
adata_PM.write(os.path.join(workdir,'adata_PM.h5ad'))
adata_PK.write(os.path.join(workdir,'adata_PK.h5ad'))
adata_C.write(os.path.join(workdir,'adata_C.h5ad'))
adata_G.write(os.path.join(workdir,'adata_G.h5ad'))
adata_P.write(os.path.join(workdir,'adata_P.h5ad'))
adata_K.write(os.path.join(workdir,'adata_K.h5ad'))
adata_M.write(os.path.join(workdir,'adata_M.h5ad'))
adata_all_CG.write(os.path.join(workdir,'adata_all_CG.h5ad'))
adata_all.write(os.path.join(workdir,'adata_all.h5ad'))
adata_all_selected.write(os.path.join(workdir,'adata_all_selected.h5ad'))
adata_cmp_CG.write(os.path.join(workdir,'adata_cmp_CG.h5ad'))
adata_cmp_CM.write(os.path.join(workdir,'adata_cmp_CM.h5ad'))
adata_cmp_CP.write(os.path.join(workdir,'adata_cmp_CP.h5ad'))
[ ]:
Read back anndata objects
adata_CG = si.read_h5ad(os.path.join(workdir,'adata_CG.h5ad'))
adata_CP = si.read_h5ad(os.path.join(workdir,'adata_CP.h5ad'))
adata_PM = si.read_h5ad(os.path.join(workdir,'adata_PM.h5ad'))
adata_PK = si.read_h5ad(os.path.join(workdir,'adata_PK.h5ad'))
adata_C = si.read_h5ad(os.path.join(workdir,'adata_C.h5ad'))
adata_G = si.read_h5ad(os.path.join(workdir,'adata_G.h5ad'))
adata_P = si.read_h5ad(os.path.join(workdir,'adata_P.h5ad'))
adata_K = si.read_h5ad(os.path.join(workdir,'adata_K.h5ad'))
adata_M = si.read_h5ad(os.path.join(workdir,'adata_M.h5ad'))
adata_all_CG = si.read_h5ad(os.path.join(workdir,'adata_all_CG.h5ad'))
adata_all = si.read_h5ad(os.path.join(workdir,'adata_all.h5ad'))
adata_all_selected = si.read_h5ad(os.path.join(workdir,'adata_all_selected.h5ad'))
adata_cmp_CG = si.read_h5ad(os.path.join(workdir,'adata_cmp_CG.h5ad'))
adata_cmp_CM = si.read_h5ad(os.path.join(workdir,'adata_cmp_CM.h5ad'))
adata_cmp_CP = si.read_h5ad(os.path.join(workdir,'adata_cmp_CP.h5ad'))
[ ]: