Multimodal gene regulatory network
Here we will use dual-omics SHARE-seq dataset, more specicially the dataset from figure4 in SHARE-seq study, “multiome_ma2020_fig4” as an example to illustrate how SIMBA performs gene regulatory network (GRN) analysis
[1]:
import os
import numpy as np
import pandas as pd
import simba as si
si.__version__
[1]:
'1.0'
[2]:
workdir = 'result_multiome_shareseq'
si.settings.set_workdir(workdir)
Saving results in: result_multiome_shareseq
[3]:
si.settings.set_figure_params(dpi=80,
style='white',
fig_size=[5,5],
rc={'image.cmap': 'viridis'})
[4]:
# to make plots prettier
from IPython.display import set_matplotlib_formats
set_matplotlib_formats('retina')
[ ]:
Identify master regulators
[5]:
adata_G = si.read_h5ad(os.path.join(workdir,'adata_G.h5ad'))
adata_M = si.read_h5ad(os.path.join(workdir,'adata_M.h5ad'))
adata_all = si.read_h5ad(os.path.join(workdir,'adata_all.h5ad'))
adata_cmp_CG = si.read_h5ad(os.path.join(workdir,'adata_cmp_CG.h5ad'))
adata_cmp_CM = si.read_h5ad(os.path.join(workdir,'adata_cmp_CM.h5ad'))
[6]:
# find paired TF motifs and TF genes
motifs_genes = pd.DataFrame(columns=['motif', 'gene'])
for x in adata_M.obs_names:
x_split = x.split('_')
for y in adata_G.obs_names:
if y in x_split:
motifs_genes.loc[motifs_genes.shape[0]] = [x,y]
[7]:
print(motifs_genes.shape)
motifs_genes.head()
(545, 2)
[7]:
| motif | gene | |
|---|---|---|
| 0 | M_ENSMUSG00000056854_LINE1392_Pou3f4_D | Pou3f4 |
| 1 | M_ENSMUSG00000039164_LINE1505_Naif1_I | Naif1 |
| 2 | M_ENSMUSG00000021779_LINE6679_Thrb_I_N2 | Thrb |
| 3 | M_ENSMUSG00000072294_LINE811_Klf12_D_N1 | Klf12 |
| 4 | M_ENSMUSG00000001288_LINE1567_Rarg_D_N1 | Rarg |
[9]:
list_tf_motif = motifs_genes['motif'].tolist()
list_tf_gene = motifs_genes['gene'].tolist()
df_metrics_motif = adata_cmp_CM.var.copy()
df_metrics_gene = adata_cmp_CG.var.copy()
[11]:
df_metrics_motif.head()
[11]:
| max | std | gini | entropy | |
|---|---|---|---|---|
| M_ENSMUSG00000045903_LINE141_Npas4_I | 1.486339 | 0.809502 | 0.390054 | 8.516953 |
| M_ENSMUSG00000056854_LINE1392_Pou3f4_D | 3.707385 | 2.825883 | 0.922120 | 5.813827 |
| M_ENSMUSG00000067261_LINE1101_Foxd3_I | 3.972934 | 3.138940 | 0.968917 | 4.672879 |
| M_ENSMUSG00000039164_LINE1505_Naif1_I | 1.641688 | 1.004522 | 0.436659 | 8.448645 |
| M_ENSMUSG00000021779_LINE6679_Thrb_I_N2 | 1.991992 | 0.862905 | 0.461154 | 8.395323 |
[12]:
df_metrics_gene.head()
[12]:
| max | std | gini | entropy | |
|---|---|---|---|---|
| Nemf | 0.953891 | 0.414289 | 0.223926 | 8.689371 |
| 4930524O07Rik | 0.770682 | 0.257394 | 0.147313 | 8.734219 |
| Zfp455 | 0.875628 | 0.287687 | 0.166179 | 8.724028 |
| Casp12 | 0.939308 | 0.323212 | 0.184197 | 8.714138 |
| Coro1a | 0.978200 | 0.403667 | 0.224001 | 8.689423 |
[ ]:
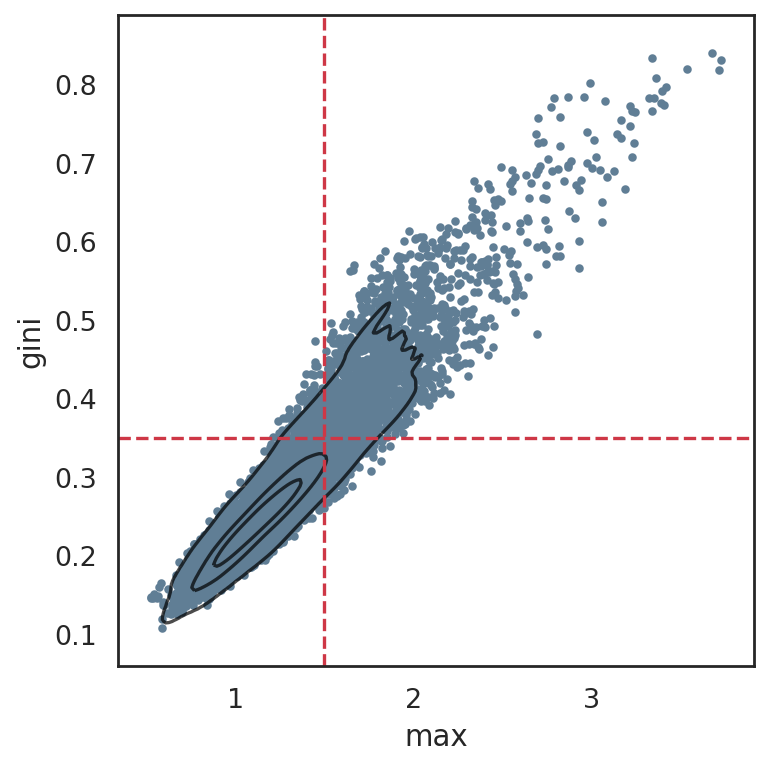
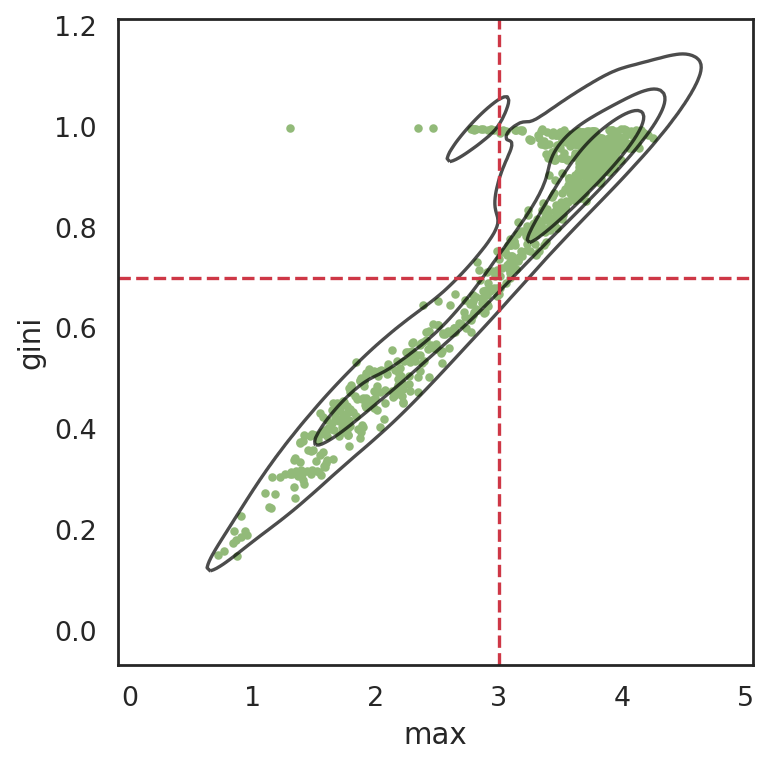
plot SIMBA metrics in order to help set the cutoffs later
[13]:
si.pl.entity_metrics(adata_cmp_CG,x='max',y='gini',
show_texts=False,
show_cutoff=True,
show_contour=True,
c='#607e95',
cutoff_x=1.5,
cutoff_y=0.35)
[14]:
si.pl.entity_metrics(adata_cmp_CM,x='max',y='gini',
show_texts=False,
show_cutoff=True,
show_contour=True,
c='#92ba79',
cutoff_x=3,
cutoff_y=0.7)
[ ]:
identify master regulators
[15]:
df_MR = si.tl.find_master_regulators(adata_all,
list_tf_motif=list_tf_motif,
list_tf_gene=list_tf_gene,
cutoff_gene_max=1.5,
cutoff_gene_gini=0.35,
cutoff_motif_max=3,
cutoff_motif_gini=0.7,
metrics_gene=df_metrics_gene,
metrics_motif=df_metrics_motif
)
/data/pinello/SHARED_SOFTWARE/anaconda_latest/envs/hc_simba/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
Adding motif metrics ...
Adding gene metrics ...
Computing distances between TF motifs and genes ...
filtering master regulators based on gene metrics:
Gini index
max
filtering master regulators based on motif metrics:
Gini index
max
[16]:
df_MR
[16]:
| motif | gene | rank | dist | max_motif | std_motif | gini_motif | entropy_motif | max_gene | std_gene | gini_gene | entropy_gene | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | M_ENSMUSG00000002983_LINE7015_Relb_I | Relb | 6.0 | 1.841419 | 4.137951 | 2.204292 | 0.958187 | 5.173000 | 2.200178 | 0.819749 | 0.475906 | 8.339973 |
| 1 | M_ENSMUSG00000027985_LINE1723_Lef1_D | Lef1 | 20.0 | 1.777754 | 3.629514 | 2.863067 | 0.896504 | 6.426489 | 1.555286 | 0.881973 | 0.448684 | 8.439009 |
| 2 | M_ENSMUSG00000005836_LINE1110_Gata6_D | Gata6 | 20.0 | 2.180344 | 4.005656 | 3.809256 | 0.968346 | 4.878388 | 2.022788 | 0.665929 | 0.412207 | 8.438866 |
| 3 | M_ENSMUSG00000025225_LINE1658_Nfkb2_I | Nfkb2 | 50.0 | 2.329539 | 3.904178 | 1.802294 | 0.922550 | 5.768632 | 1.762300 | 0.665998 | 0.375743 | 8.521793 |
| 4 | M_ENSMUSG00000033016_LINE1662_Nfatc1_I | Nfatc1 | 54.0 | 2.451544 | 3.552284 | 2.898845 | 0.977447 | 3.644386 | 1.787450 | 0.620042 | 0.365021 | 8.528356 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 63 | M_ENSMUSG00000026077_LINE84_Npas2_D | Npas2 | 17207.0 | 4.290294 | 3.382352 | 2.224144 | 0.946492 | 4.167462 | 2.068159 | 0.851327 | 0.467876 | 8.372712 |
| 64 | M_ENSMUSG00000028940_LINE94_Hes2_D | Hes2 | 17221.0 | 3.984124 | 3.309705 | 1.710030 | 0.817412 | 6.702363 | 1.796419 | 0.752017 | 0.428637 | 8.447193 |
| 65 | M_ENSMUSG00000042745_LINE133_Id1_I | Id1 | 17257.0 | 4.314670 | 3.443919 | 2.221636 | 0.934407 | 4.687283 | 1.907240 | 0.794275 | 0.432841 | 8.438908 |
| 66 | M_ENSMUSG00000038415_LINE1071_Foxq1_I | Foxq1 | 17321.0 | 2.686434 | 3.650490 | 2.118908 | 0.890306 | 6.075940 | 2.636956 | 1.002889 | 0.630324 | 7.898009 |
| 67 | M_ENSMUSG00000038151_LINE465_Prdm1_I | Prdm1 | 17352.0 | 3.679676 | 3.006260 | 1.422200 | 0.703327 | 7.631708 | 2.698913 | 1.290094 | 0.725242 | 7.627998 |
68 rows × 12 columns
[ ]:
Identify target genes
[ ]:
[20]:
adata_CP = si.read_h5ad(os.path.join(workdir,'adata_CP.h5ad'))
adata_PM = si.read_h5ad(os.path.join(workdir,'adata_PM.h5ad'))
#make sure the indices of motifs are the same as in `motifs_genes`
adata_PM.var.index = 'M_' + adata_PM.var.index
[21]:
master_regulators = ['Lef1', 'Hoxc13', 'Relb', 'Gata6', 'Nfatc1']
list_tf_motif = motifs_genes[np.isin(motifs_genes['gene'], master_regulators)]['motif'].tolist()
list_tf_gene = motifs_genes[np.isin(motifs_genes['gene'], master_regulators)]['gene'].tolist()
[ ]:
The following step can be ignored if si.tl.gene_scores(adata_CP) has been performed before
[22]:
# get the overlap between peaks and gene loci
# the field `uns['gene_scores']['overlap']` will be added to adata_CP
_ = si.tl.gene_scores(adata_CP,genome='mm10',use_gene_weigt=True,use_top_pcs=False)
Gene scores are being calculated for the first time
`use_precomputed` has been ignored
Processing: 0.0%
Processing: 20.0%
Processing: 40.0%
Processing: 60.0%
Processing: 80.0%
/data/pinello/SHARED_SOFTWARE/anaconda_latest/envs/hc_simba/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
[23]:
dict_tf_targets = si.tl.find_target_genes(adata_all,
adata_PM,
list_tf_motif=list_tf_motif,
list_tf_gene=list_tf_gene,
adata_CP=adata_CP,
cutoff_gene=5000)
Preprocessing ...
/data/pinello/SHARED_SOFTWARE/anaconda_latest/envs/hc_simba/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
#genes: 17399
#peaks: 332987
#genes-associated peaks: 318851
computing distances between genes and genes-associated peaks ...
Saving variables into `.uns['tf_targets']` ...
searching for target genes of M_ENSMUSG00000005836_LINE1110_Gata6_D
#candinate genes is 400
removing duplicate genes ...
removing genes that do not contain TF motif ...
#candinate genes is 213
completed: 0.0%
completed: 19.7%
completed: 39.4%
completed: 59.2%
completed: 78.9%
completed: 98.6%
Pruning candidate genes based on nearby peaks ...
Pruning candidate genes based on average rank ...
searching for target genes of M_ENSMUSG00000033016_LINE1662_Nfatc1_I
/data/pinello/SHARED_SOFTWARE/anaconda_latest/envs/hc_simba/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
#candinate genes is 400
removing duplicate genes ...
removing genes that do not contain TF motif ...
#candinate genes is 232
completed: 0.0%
completed: 19.8%
completed: 39.7%
completed: 59.5%
completed: 79.3%
completed: 99.1%
Pruning candidate genes based on nearby peaks ...
Pruning candidate genes based on average rank ...
searching for target genes of M_ENSMUSG00000027985_LINE1723_Lef1_D
/data/pinello/SHARED_SOFTWARE/anaconda_latest/envs/hc_simba/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
#candinate genes is 400
removing duplicate genes ...
removing genes that do not contain TF motif ...
#candinate genes is 289
completed: 0.0%
completed: 19.7%
completed: 39.4%
completed: 59.2%
completed: 78.9%
completed: 98.6%
Pruning candidate genes based on nearby peaks ...
Pruning candidate genes based on average rank ...
searching for target genes of M_ENSMUSG00000002983_LINE7015_Relb_I
/data/pinello/SHARED_SOFTWARE/anaconda_latest/envs/hc_simba/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
#candinate genes is 400
removing duplicate genes ...
removing genes that do not contain TF motif ...
#candinate genes is 226
completed: 0.0%
completed: 19.9%
completed: 39.8%
completed: 59.7%
completed: 79.6%
completed: 99.6%
Pruning candidate genes based on nearby peaks ...
Pruning candidate genes based on average rank ...
searching for target genes of M_ENSMUSG00000001655_LINE1151_Hoxc13_D
/data/pinello/SHARED_SOFTWARE/anaconda_latest/envs/hc_simba/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
#candinate genes is 400
removing duplicate genes ...
removing genes that do not contain TF motif ...
#candinate genes is 259
completed: 0.0%
completed: 19.7%
completed: 39.4%
completed: 59.1%
completed: 78.8%
completed: 98.5%
Pruning candidate genes based on nearby peaks ...
Pruning candidate genes based on average rank ...
[ ]:
[24]:
dict_tf_targets.keys()
[24]:
dict_keys(['M_ENSMUSG00000005836_LINE1110_Gata6_D', 'M_ENSMUSG00000033016_LINE1662_Nfatc1_I', 'M_ENSMUSG00000027985_LINE1723_Lef1_D', 'M_ENSMUSG00000002983_LINE7015_Relb_I', 'M_ENSMUSG00000001655_LINE1151_Hoxc13_D'])
[25]:
dict_tf_targets['M_ENSMUSG00000027985_LINE1723_Lef1_D']
[25]:
| average_rank | has_motif | rank_gene_to_TFmotif | rank_gene_to_TFgene | rank_peak_to_TFmotif | rank_peak2_to_TFmotif | rank_peak_to_gene | rank_peak2_to_gene | |
|---|---|---|---|---|---|---|---|---|
| Slc16a6 | 10.0 | yes | 2.0 | 18.0 | 2364.0 | 13824.0 | 917.0 | 917.0 |
| Lef1 | 10.5 | yes | 20.0 | 1.0 | 121.0 | 2150.0 | 803.0 | 803.0 |
| Psd3 | 14.0 | yes | 26.0 | 2.0 | 181.0 | 3591.0 | 522.0 | 522.0 |
| Tle4 | 17.5 | yes | 31.0 | 4.0 | 2.0 | 2.0 | 26.0 | 26.0 |
| Tiam2 | 18.0 | yes | 25.0 | 11.0 | 69.0 | 69.0 | 654.0 | 654.0 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... |
| Cldn10 | 3857.0 | yes | 68.0 | 7646.0 | 1462.0 | 121799.0 | 59.0 | 59.0 |
| Has3 | 4011.0 | yes | 35.0 | 7987.0 | 3271.0 | 26782.0 | 723.0 | 723.0 |
| Npas2 | 4300.0 | yes | 147.0 | 8453.0 | 141.0 | 220013.0 | 1301.0 | 1301.0 |
| Rgs9 | 4346.5 | yes | 37.0 | 8656.0 | 669.0 | 27978.0 | 118.0 | 118.0 |
| Msx1 | 4902.5 | yes | 193.0 | 9612.0 | 71.0 | 1127.0 | 271.0 | 271.0 |
171 rows × 8 columns
[26]:
dict_tf_targets['M_ENSMUSG00000001655_LINE1151_Hoxc13_D']
[26]:
| average_rank | has_motif | rank_gene_to_TFmotif | rank_gene_to_TFgene | rank_peak_to_TFmotif | rank_peak2_to_TFmotif | rank_peak_to_gene | rank_peak2_to_gene | |
|---|---|---|---|---|---|---|---|---|
| Cybrd1 | 43.5 | yes | 66.0 | 21.0 | 341.0 | 341.0 | 1228.0 | 1228.0 |
| Tiam2 | 47.0 | yes | 87.0 | 7.0 | 3496.0 | 5221.0 | 654.0 | 654.0 |
| Sema4c | 64.5 | yes | 35.0 | 94.0 | 79.0 | 72730.0 | 102.0 | 102.0 |
| Ammecr1 | 65.5 | yes | 57.0 | 74.0 | 1777.0 | 2668.0 | 760.0 | 760.0 |
| Pim1 | 66.0 | yes | 108.0 | 24.0 | 682.0 | 43191.0 | 4908.0 | 4908.0 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... |
| Dapl1 | 3125.5 | yes | 19.0 | 6232.0 | 359.0 | 359.0 | 227.0 | 227.0 |
| Vipr1 | 3335.5 | yes | 20.0 | 6651.0 | 975.0 | 18759.0 | 825.0 | 825.0 |
| Myo5c | 3809.5 | yes | 84.0 | 7535.0 | 807.0 | 807.0 | 19.0 | 19.0 |
| Krt81 | 3872.0 | yes | 7.0 | 7737.0 | 7.0 | 38.0 | 3.0 | 3.0 |
| Hspb8 | 4655.5 | yes | 40.0 | 9271.0 | 909.0 | 19478.0 | 1383.0 | 1383.0 |
133 rows × 8 columns
[ ]:
save results
[27]:
adata_CP.write(os.path.join(workdir,'adata_CP.h5ad'))
[28]:
import pickle
with open('./result_multiome_shareseq/dict_tf_targets.pickle', 'wb') as handle:
pickle.dump(dict_tf_targets, handle, protocol=pickle.HIGHEST_PROTOCOL)
Read back pickle file
with open('./result_multiome_shareseq/dict_tf_targets.pickle', 'rb') as handle:
dict_tf_targets = pickle.load(handle)
[ ]: