Batch correction (two batches)
Here we will use two scRNA-seq mouse atlas datasets of different technologies as an example to illustrate how SIMBA performs scRNA-seq batch correction
[1]:
import os
import simba as si
si.__version__
[1]:
'1.0'
[2]:
workdir = 'result_mouse_atlas'
si.settings.set_workdir(workdir)
Saving results in: result_mouse_atlas
[3]:
si.settings.set_figure_params(dpi=80,
style='white',
fig_size=[5,5],
rc={'image.cmap': 'viridis'})
[4]:
# to make plots prettier
from IPython.display import set_matplotlib_formats
set_matplotlib_formats('retina')
[ ]:
load example data
[5]:
# mirowell-seq data
adata_CG_mi = si.datasets.rna_han2018()
rna_han2018.h5ad: 0.00B [00:00, ?B/s]
Downloading data ...
rna_han2018.h5ad: 24.6MB [00:17, 1.45MB/s]
Downloaded to result_mouse_atlas/data.
[6]:
# smart-seq data
adata_CG_sm = si.datasets.rna_tmc2018()
rna_tmc2018.h5ad: 0.00B [00:00, ?B/s]
Downloading data ...
rna_tmc2018.h5ad: 50.9MB [00:31, 1.60MB/s]
Downloaded to result_mouse_atlas/data.
[7]:
adata_CG_mi
[7]:
AnnData object with n_obs × n_vars = 4239 × 15006
obs: 'nGene', 'nUMI', 'orig.ident', 'batch', 'ct', 'ct.orig', 'tissue', 'organ', 'percent.mito', 'batchlb', 'celltype'
[8]:
adata_CG_sm
[8]:
AnnData object with n_obs × n_vars = 2715 × 15006
obs: 'nGene', 'nUMI', 'orig.ident', 'batch', 'ct', 'ct.orig', 'tissue', 'organ', 'percent.mito', 'batchlb', 'celltype'
[ ]:
microwell-seq data
preprocessing
[9]:
si.pp.filter_genes(adata_CG_mi,min_n_cells=3)
Before filtering:
4239 cells, 15006 genes
Filter genes based on min_n_cells
After filtering out low-expressed genes:
4239 cells, 13662 genes
/data/pinello/SHARED_SOFTWARE/anaconda_latest/envs/hc_simba/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
[10]:
si.pp.cal_qc_rna(adata_CG_mi)
[11]:
si.pl.violin(adata_CG_mi,list_obs=['n_counts','n_genes','pct_mt'])
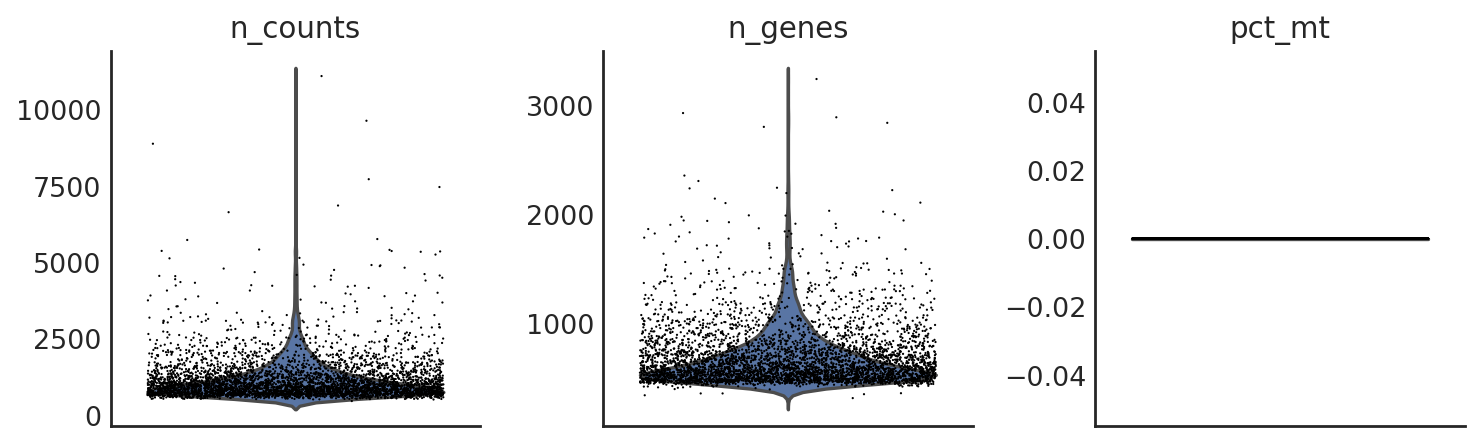
[12]:
si.pp.normalize(adata_CG_mi,method='lib_size')
[13]:
si.pp.log_transform(adata_CG_mi)
[14]:
si.pp.select_variable_genes(adata_CG_mi, n_top_genes=3000)
3000 variable genes are selected.
[15]:
si.pl.variable_genes(adata_CG_mi,show_texts=True,)
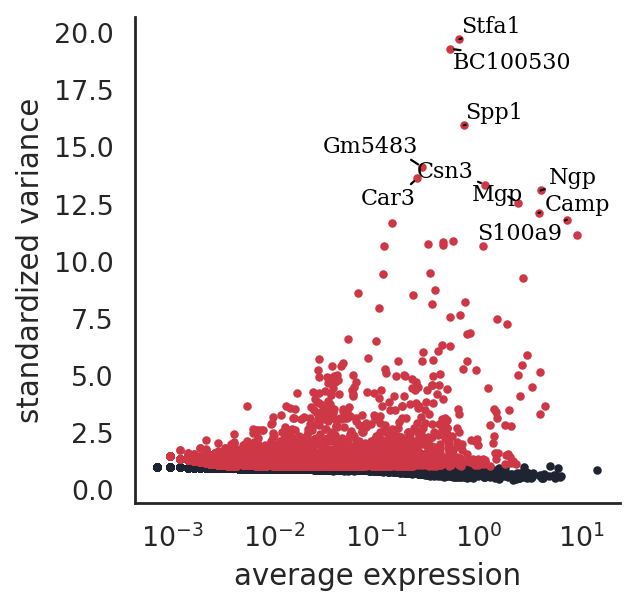
discretize RNA expression
[16]:
si.tl.discretize(adata_CG_mi,n_bins=5)
si.pl.discretize(adata_CG_mi,kde=False)
[0.64202917 1.8883792 2.4364908 3.030551 3.853816 7.666325 ]
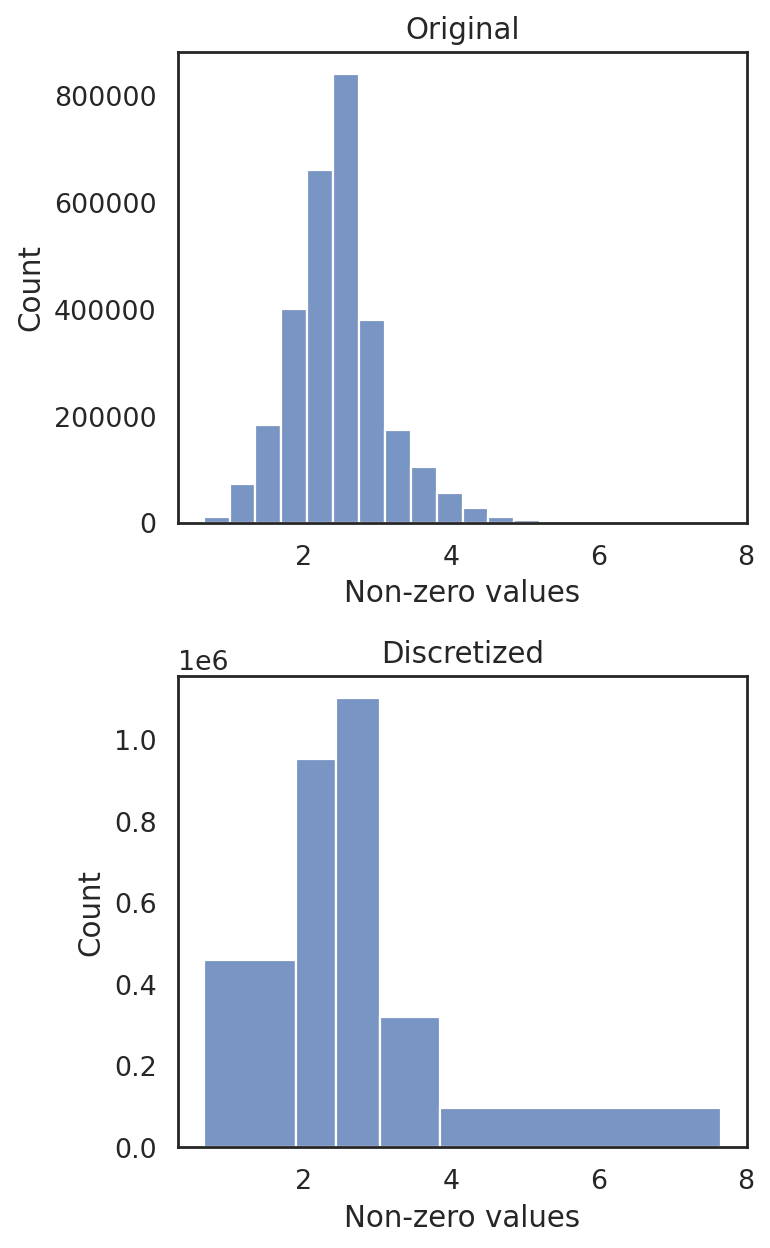
[ ]:
smart-seq2 data
preprocessing
[17]:
si.pp.filter_genes(adata_CG_sm,min_n_cells=3)
Before filtering:
2715 cells, 15006 genes
Filter genes based on min_n_cells
After filtering out low-expressed genes:
2715 cells, 14956 genes
/data/pinello/SHARED_SOFTWARE/anaconda_latest/envs/hc_simba/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
[18]:
si.pp.cal_qc_rna(adata_CG_sm)
[19]:
si.pl.violin(adata_CG_sm,list_obs=['n_counts','n_genes','pct_mt'])
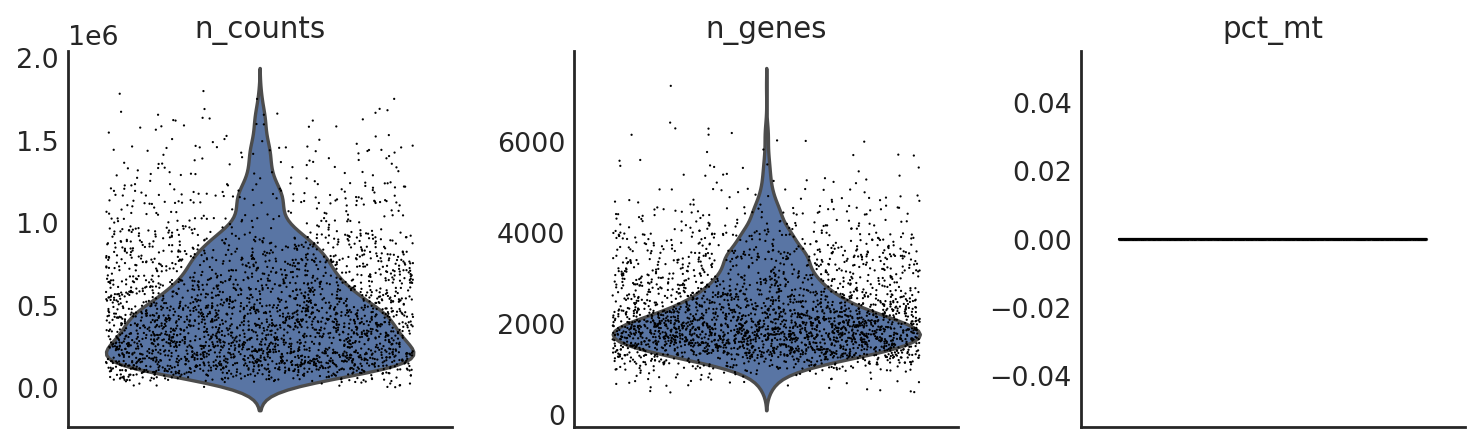
[20]:
si.pp.normalize(adata_CG_sm,method='lib_size')
[21]:
si.pp.log_transform(adata_CG_sm)
[22]:
si.pp.select_variable_genes(adata_CG_sm, n_top_genes=3000)
3000 variable genes are selected.
[23]:
si.pl.variable_genes(adata_CG_sm,show_texts=True,)
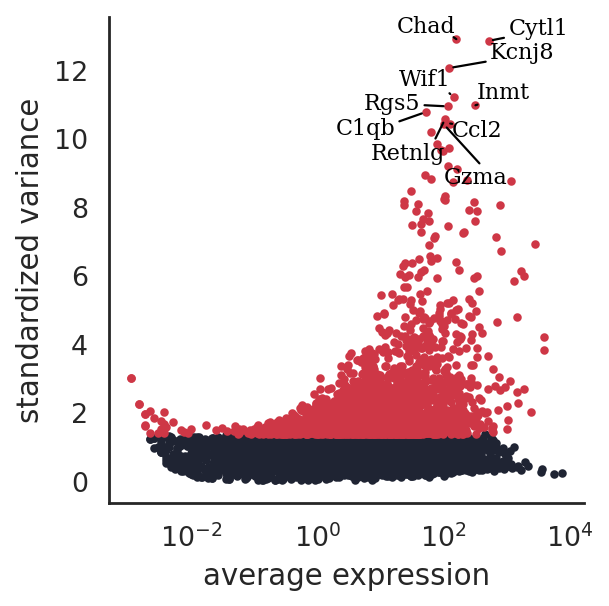
discretize RNA expression
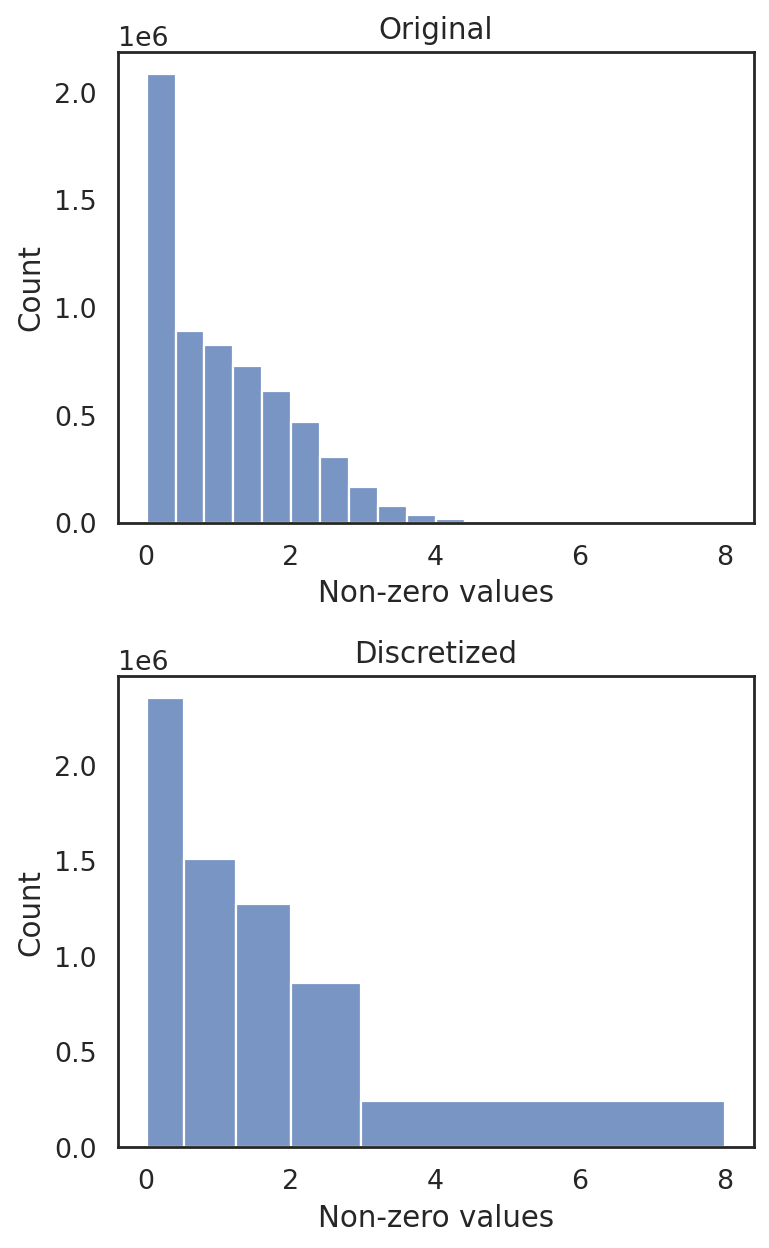
[24]:
si.tl.discretize(adata_CG_sm,n_bins=5)
si.pl.discretize(adata_CG_sm,kde=False)
[5.5492530e-03 5.2105367e-01 1.2380612e+00 2.0028372e+00 2.9648428e+00
8.0040236e+00]
[ ]:
infer edges between cells of different technologies
[25]:
adata_CmiCsm = si.tl.infer_edges(adata_CG_mi, adata_CG_sm, n_components=20, k=20)
#shared features: 2969
Performing randomized SVD ...
/data/pinello/SHARED_SOFTWARE/anaconda_latest/envs/hc_simba/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
Searching for mutual nearest neighbors ...
27953 edges are selected
[26]:
adata_CmiCsm
[26]:
AnnData object with n_obs × n_vars = 4239 × 2715
obs: 'nGene', 'nUMI', 'orig.ident', 'batch', 'ct', 'ct.orig', 'tissue', 'organ', 'percent.mito', 'batchlb', 'celltype', 'n_counts', 'n_genes', 'pct_genes', 'pct_mt'
var: 'nGene', 'nUMI', 'orig.ident', 'batch', 'ct', 'ct.orig', 'tissue', 'organ', 'percent.mito', 'batchlb', 'celltype', 'n_counts', 'n_genes', 'pct_genes', 'pct_mt'
obsm: 'svd'
varm: 'svd'
layers: 'conn'
[27]:
si.pl.node_similarity(adata_CmiCsm, cutoff=0.5)
#selected edges: 27953
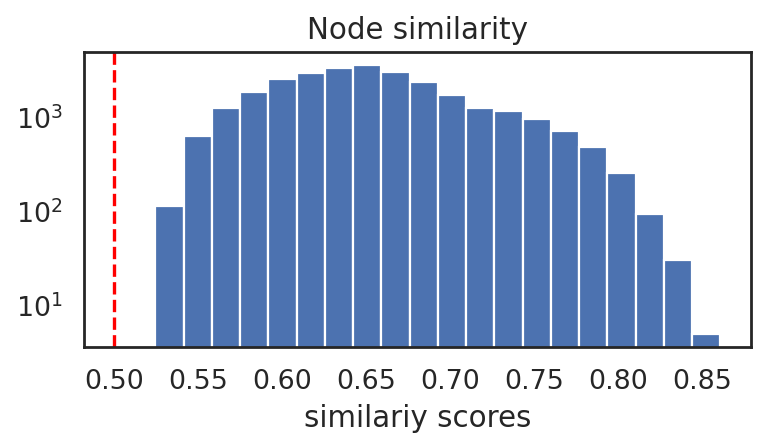
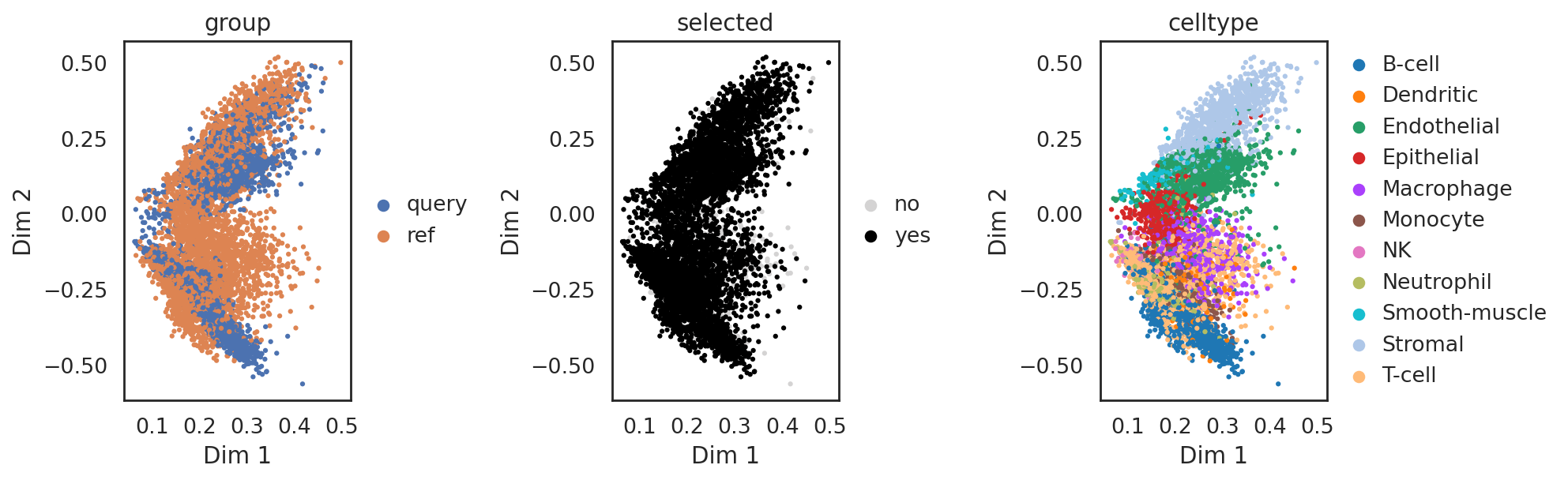
[28]:
si.pl.svd_nodes(adata_CmiCsm,color=['celltype'],cutoff=0.5)
[29]:
# edges can be futhere trimmed if needed. Here we keep all of them
si.tl.trim_edges(adata_CmiCsm, cutoff=0.5)
27953 edges are selected
[ ]:
generate graph
[30]:
si.tl.gen_graph(list_CG=[adata_CG_mi, adata_CG_sm],
list_CC=[adata_CmiCsm],
copy=False,
use_highly_variable=True,
dirname='graph0')
/data/pinello/SHARED_SOFTWARE/anaconda_latest/envs/hc_simba/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
relation0: source: C, destination: G
#edges: 67106
relation1: source: C, destination: G
#edges: 165204
relation2: source: C, destination: G
#edges: 208293
relation3: source: C, destination: G
#edges: 79190
relation4: source: C, destination: G
#edges: 30704
relation5: source: C2, destination: G
#edges: 289468
relation6: source: C2, destination: G
#edges: 130323
relation7: source: C2, destination: G
#edges: 137814
relation8: source: C2, destination: G
#edges: 138614
relation9: source: C2, destination: G
#edges: 77368
relation10: source: C, destination: C2
#edges: 27953
Total number of edges: 1352037
Writing graph file "pbg_graph.txt" to "result_mouse_atlas/pbg/graph0" ...
Finished.
[ ]:
PBG training
Before PBG training, let’s take a look at the parameters:
[31]:
si.settings.pbg_params
[31]:
{'entity_path': 'result_mouse_atlas/pbg/graph0/input/entity',
'edge_paths': ['result_mouse_atlas/pbg/graph0/input/edge'],
'checkpoint_path': '',
'entities': {'C': {'num_partitions': 1},
'C2': {'num_partitions': 1},
'G': {'num_partitions': 1}},
'relations': [{'name': 'r0',
'lhs': 'C',
'rhs': 'G',
'operator': 'none',
'weight': 1.0},
{'name': 'r1', 'lhs': 'C', 'rhs': 'G', 'operator': 'none', 'weight': 2.0},
{'name': 'r2', 'lhs': 'C', 'rhs': 'G', 'operator': 'none', 'weight': 3.0},
{'name': 'r3', 'lhs': 'C', 'rhs': 'G', 'operator': 'none', 'weight': 4.0},
{'name': 'r4', 'lhs': 'C', 'rhs': 'G', 'operator': 'none', 'weight': 5.0},
{'name': 'r5', 'lhs': 'C2', 'rhs': 'G', 'operator': 'none', 'weight': 1.0},
{'name': 'r6', 'lhs': 'C2', 'rhs': 'G', 'operator': 'none', 'weight': 2.0},
{'name': 'r7', 'lhs': 'C2', 'rhs': 'G', 'operator': 'none', 'weight': 3.0},
{'name': 'r8', 'lhs': 'C2', 'rhs': 'G', 'operator': 'none', 'weight': 4.0},
{'name': 'r9', 'lhs': 'C2', 'rhs': 'G', 'operator': 'none', 'weight': 5.0},
{'name': 'r10',
'lhs': 'C',
'rhs': 'C2',
'operator': 'none',
'weight': 10.0}],
'dynamic_relations': False,
'dimension': 50,
'global_emb': False,
'comparator': 'dot',
'num_epochs': 10,
'workers': 4,
'num_batch_negs': 50,
'num_uniform_negs': 50,
'loss_fn': 'softmax',
'lr': 0.1,
'early_stopping': False,
'regularization_coef': 0.0,
'wd': 0.0,
'wd_interval': 50,
'eval_fraction': 0.05,
'eval_num_batch_negs': 50,
'eval_num_uniform_negs': 50,
'checkpoint_preservation_interval': None}
[ ]:
If no parameters need to be adjusted, the training can be simply done with:
si.tl.pbg_train(auto_wd=True, save_wd=True, output='model')
[ ]:
Here we show how to adjust training-related parameters if needed. In general, weight decay wd is the only parameter that might need to be adjusted based on the following pbg metric plots. However, in almost all the cases, the automatically decided wd (enabling it by setting auto_wd=True) works well.
E.g. we want to change the number of cpus workers:
[32]:
# modify parameters
dict_config = si.settings.pbg_params.copy()
# dict_config['wd'] = 0.026209
dict_config['workers'] = 12
## start training
si.tl.pbg_train(pbg_params = dict_config, auto_wd=True, save_wd=True, output='model')
Auto-estimated weight decay is 0.026209
`.settings.pbg_params['wd']` has been updated to 0.026209
Converting input data ...
[2021-06-29 19:27:45.271681] Using the 11 relation types given in the config
[2021-06-29 19:27:45.272150] Searching for the entities in the edge files...
[2021-06-29 19:27:47.088293] Entity type C:
[2021-06-29 19:27:47.088849] - Found 4239 entities
[2021-06-29 19:27:47.089114] - Removing the ones with fewer than 1 occurrences...
[2021-06-29 19:27:47.089864] - Left with 4239 entities
[2021-06-29 19:27:47.090766] - Shuffling them...
[2021-06-29 19:27:47.093503] Entity type C2:
[2021-06-29 19:27:47.093749] - Found 2715 entities
[2021-06-29 19:27:47.093985] - Removing the ones with fewer than 1 occurrences...
[2021-06-29 19:27:47.094526] - Left with 2715 entities
[2021-06-29 19:27:47.094767] - Shuffling them...
[2021-06-29 19:27:47.096571] Entity type G:
[2021-06-29 19:27:47.096819] - Found 4620 entities
[2021-06-29 19:27:47.097059] - Removing the ones with fewer than 1 occurrences...
[2021-06-29 19:27:47.097742] - Left with 4620 entities
[2021-06-29 19:27:47.098011] - Shuffling them...
[2021-06-29 19:27:47.100944] Preparing counts and dictionaries for entities and relation types:
[2021-06-29 19:27:47.118889] - Writing count of entity type C and partition 0
[2021-06-29 19:27:47.132935] - Writing count of entity type C2 and partition 0
[2021-06-29 19:27:47.144771] - Writing count of entity type G and partition 0
[2021-06-29 19:27:47.156363] Preparing edge path result_mouse_atlas/pbg/graph0/input/edge, out of the edges found in result_mouse_atlas/pbg/graph0/pbg_graph.txt
using fast version
[2021-06-29 19:27:47.160494] Taking the fast train!
[2021-06-29 19:27:47.662928] - Processed 100000 edges so far...
[2021-06-29 19:27:48.158937] - Processed 200000 edges so far...
[2021-06-29 19:27:48.652791] - Processed 300000 edges so far...
[2021-06-29 19:27:49.150010] - Processed 400000 edges so far...
[2021-06-29 19:27:49.645138] - Processed 500000 edges so far...
[2021-06-29 19:27:50.140336] - Processed 600000 edges so far...
[2021-06-29 19:27:50.640567] - Processed 700000 edges so far...
[2021-06-29 19:27:51.137272] - Processed 800000 edges so far...
[2021-06-29 19:27:51.633798] - Processed 900000 edges so far...
[2021-06-29 19:27:52.130197] - Processed 1000000 edges so far...
[2021-06-29 19:27:52.627141] - Processed 1100000 edges so far...
[2021-06-29 19:27:53.124314] - Processed 1200000 edges so far...
[2021-06-29 19:27:53.619820] - Processed 1300000 edges so far...
[2021-06-29 19:27:57.876732] - Processed 1352037 edges in total
Starting training ...
Finished
[ ]:
If
wdis specified by users instead of being automatically decided, then make sure to update it in simba setting:
si.settings.pbg_params = dict_config.copy()
[ ]:
The trained result can be loaded in with the following steps:
By default, it’s using the current training result stored in .setting.pbg_params
# load in graph ('graph0') info
si.load_graph_stats()
# load in model info for ('graph0')
si.load_pbg_config()
Users can also specify different pathss
# load in graph ('graph0') info
si.load_graph_stats(path='./result_mouse_atlas/pbg/graph0/')
# load in model info for ('graph0')
si.load_pbg_config(path='./result_mouse_atlas/pbg/graph0/model/')
[ ]:
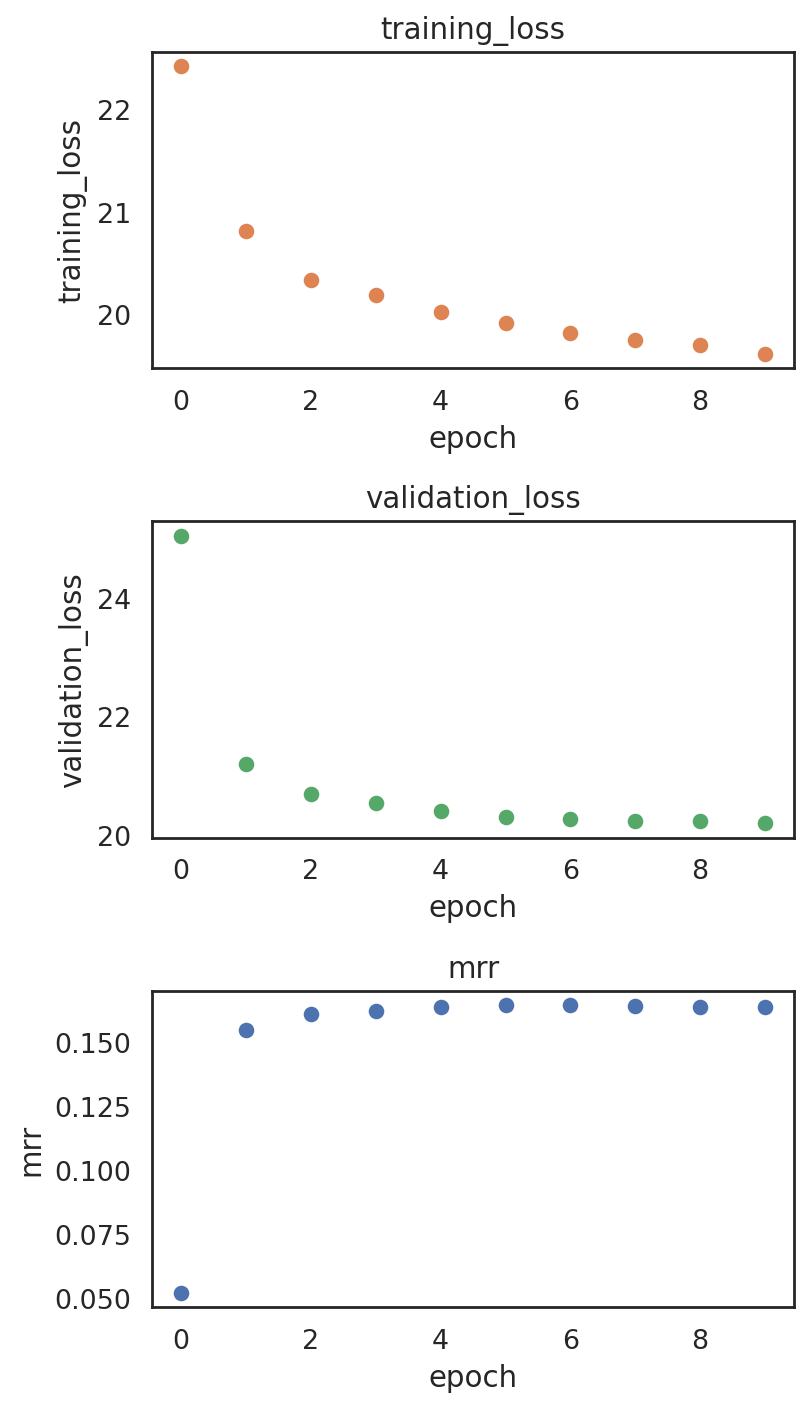
Plotting training metrics to make sure the model is not overfitting
[33]:
si.pl.pbg_metrics(fig_ncol=1)
[ ]:
Post-training Analysis
[34]:
dict_adata = si.read_embedding()
[35]:
dict_adata
[35]:
{'C': AnnData object with n_obs × n_vars = 4239 × 50,
'C2': AnnData object with n_obs × n_vars = 2715 × 50,
'G': AnnData object with n_obs × n_vars = 4620 × 50}
[36]:
adata_C = dict_adata['C'] # embeddings for cells
adata_C2 = dict_adata['C2'] # embeddings for C2
adata_G = dict_adata['G'] # embeddings for genes
[37]:
adata_C
[37]:
AnnData object with n_obs × n_vars = 4239 × 50
[38]:
adata_C2
[38]:
AnnData object with n_obs × n_vars = 2715 × 50
[39]:
adata_G
[39]:
AnnData object with n_obs × n_vars = 4620 × 50
[ ]:
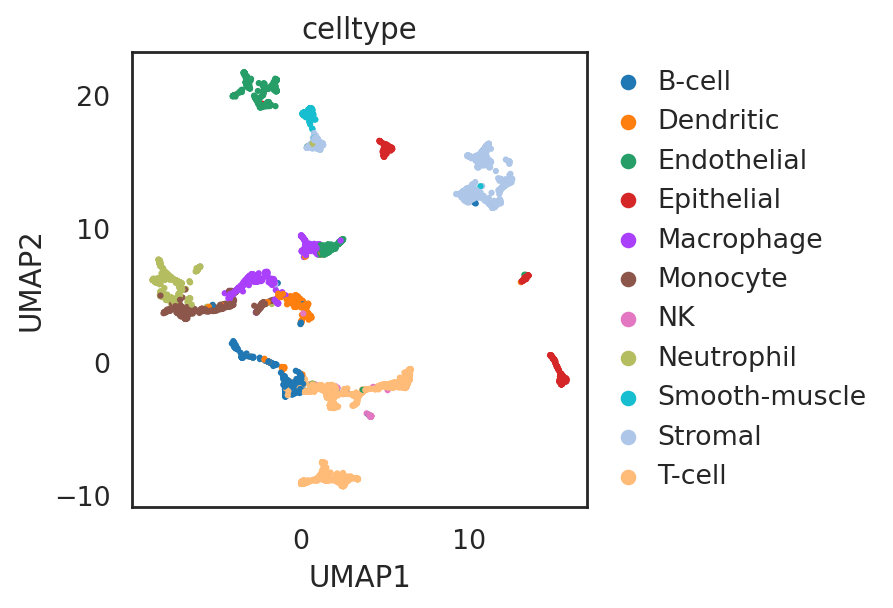
visualize embeddings of cells (microwell-seq)
[40]:
## Add annotation of celltypes (optional)
adata_C.obs['celltype'] = adata_CG_mi[adata_C.obs_names,:].obs['celltype'].copy()
adata_C
[40]:
AnnData object with n_obs × n_vars = 4239 × 50
obs: 'celltype'
[41]:
si.tl.umap(adata_C,n_neighbors=15,n_components=2)
[42]:
si.pl.umap(adata_C,
color=['celltype'],
fig_size=(5.5, 4),
drawing_order='random')
[ ]:
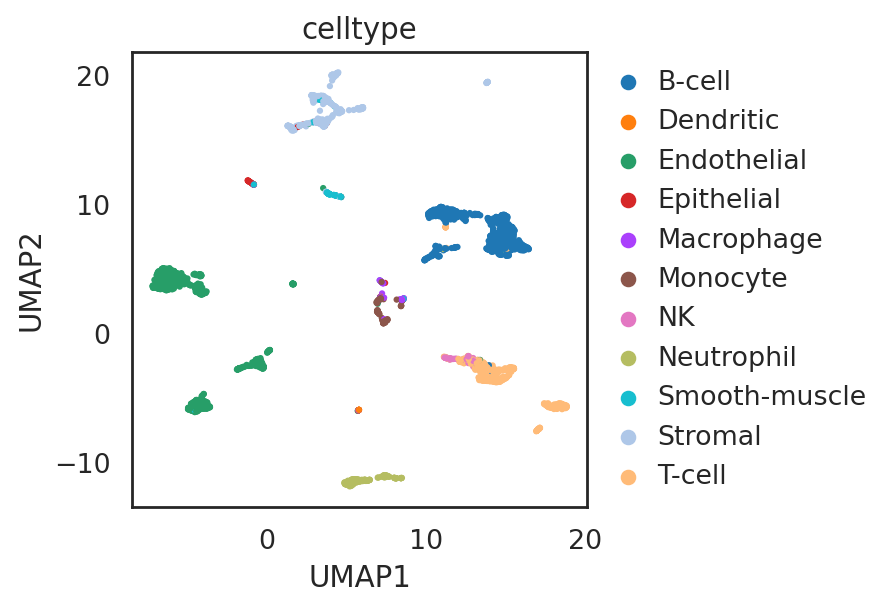
visualize embeddings of cells (smart-seq2)
[43]:
## Add annotation of celltypes (optional)
adata_C2.obs['celltype'] = adata_CG_sm[adata_C2.obs_names,:].obs['celltype'].copy()
adata_C2
/data/pinello/SHARED_SOFTWARE/anaconda_latest/envs/hc_simba/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
[43]:
AnnData object with n_obs × n_vars = 2715 × 50
obs: 'celltype'
[44]:
si.tl.umap(adata_C2,n_neighbors=15,n_components=2)
[45]:
si.pl.umap(adata_C2,
color=['celltype'],
fig_size=(5.5, 4),
drawing_order='random')
[ ]:
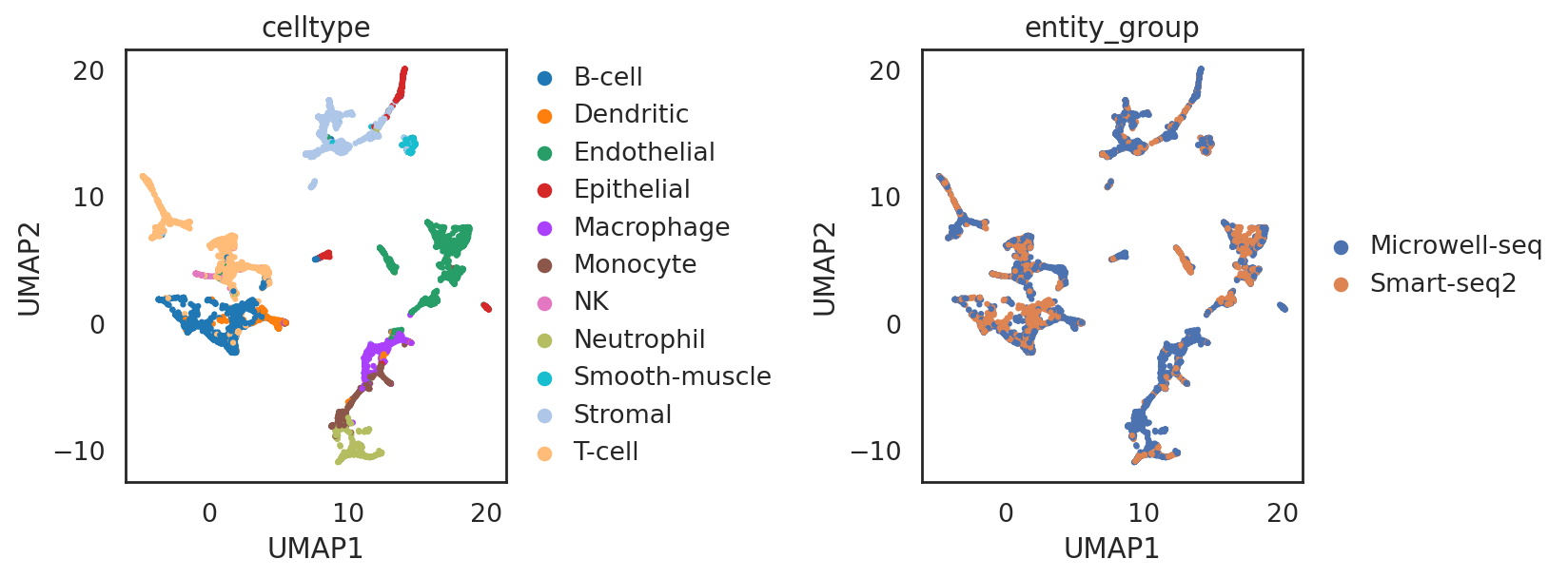
visualize co-embeddings of two batches
[46]:
adata_all = si.tl.embed(adata_ref=adata_C,list_adata_query=[adata_C2])
Performing softmax transformation for query data 0;
[47]:
## add annotations of two batches
adata_all.obs['entity_group'] = ""
adata_all.obs.loc[adata_C.obs_names, 'entity_group'] = "Microwell-seq"
adata_all.obs.loc[adata_C2.obs_names, 'entity_group'] = "Smart-seq2"
adata_all.obs.head()
[47]:
| celltype | id_dataset | entity_group | |
|---|---|---|---|
| Stromal_Pancreas_1.AAGTACATTCCATCTACC | Stromal | ref | Microwell-seq |
| Neutrophil_FetalLiver_1.GCTGTGGAATTAACCTGA | Neutrophil | ref | Microwell-seq |
| Dendritic_Kidney_2.CCTAGACAAAGTGAGGAG | Dendritic | ref | Microwell-seq |
| Endothelial_Liver_1.TGAAGCCAACAAAACCTA | Endothelial | ref | Microwell-seq |
| Stromal_Pancreas_1.CCGCTATCTACCGGGTTT | Stromal | ref | Microwell-seq |
[48]:
si.tl.umap(adata_all,n_neighbors=15,n_components=2)
[49]:
si.pl.umap(adata_all,color=['celltype','entity_group'],
drawing_order='random',
fig_size=(5,4))
[ ]:
visualize embeddings of cells and genes
[50]:
adata_all_CG = si.tl.embed(adata_ref=adata_C,
list_adata_query=[adata_C2, adata_G],
use_precomputed=False)
Performing softmax transformation for query data 0;
Performing softmax transformation for query data 1;
[51]:
## add annotations of all entities
adata_all_CG.obs['entity_anno'] = ""
adata_all_CG.obs.loc[adata_C.obs_names, 'entity_anno'] = adata_all_CG.obs.loc[adata_C.obs_names, 'celltype'].tolist()
adata_all_CG.obs.loc[adata_C2.obs_names, 'entity_anno'] = adata_all_CG.obs.loc[adata_C2.obs_names, 'celltype'].tolist()
adata_all_CG.obs.loc[adata_G.obs_names, 'entity_anno'] = 'gene'
adata_all_CG.obs.head()
[51]:
| celltype | id_dataset | entity_anno | |
|---|---|---|---|
| Stromal_Pancreas_1.AAGTACATTCCATCTACC | Stromal | ref | Stromal |
| Neutrophil_FetalLiver_1.GCTGTGGAATTAACCTGA | Neutrophil | ref | Neutrophil |
| Dendritic_Kidney_2.CCTAGACAAAGTGAGGAG | Dendritic | ref | Dendritic |
| Endothelial_Liver_1.TGAAGCCAACAAAACCTA | Endothelial | ref | Endothelial |
| Stromal_Pancreas_1.CCGCTATCTACCGGGTTT | Stromal | ref | Stromal |
[52]:
si.tl.umap(adata_all_CG,n_neighbors=15,n_components=2)
[53]:
palette_celltype = {'B-cell': '#1f77b4',
'Dendritic': '#ff7f0e',
'Endothelial': '#279e68',
'Epithelial': '#d62728',
'Macrophage': '#aa40fc',
'Monocyte': '#8c564b',
'NK': '#e377c2',
'Neutrophil': '#b5bd61',
'Smooth-muscle': '#17becf',
'Stromal': '#aec7e8',
'T-cell': '#ffbb78'}
palette_entity_anno = palette_celltype.copy()
palette_entity_anno['gene'] = "#607e95"
[54]:
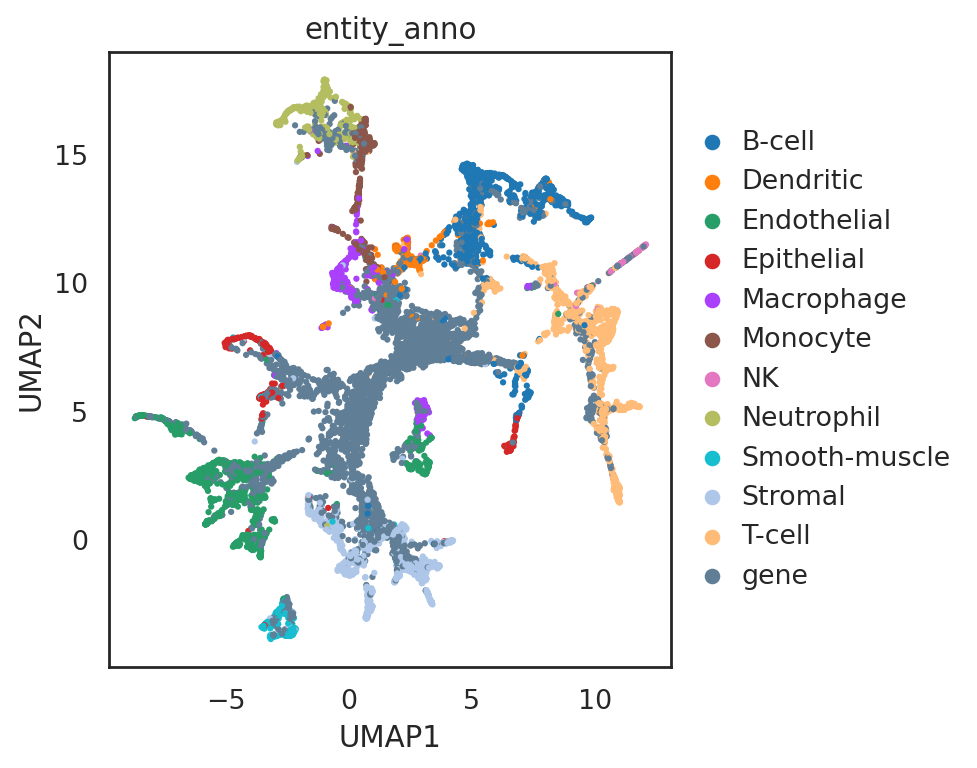
si.pl.umap(adata_all_CG,
color=['entity_anno'],
dict_palette={'entity_anno': palette_entity_anno},
drawing_order='random',
fig_size=(6,5))
[55]:
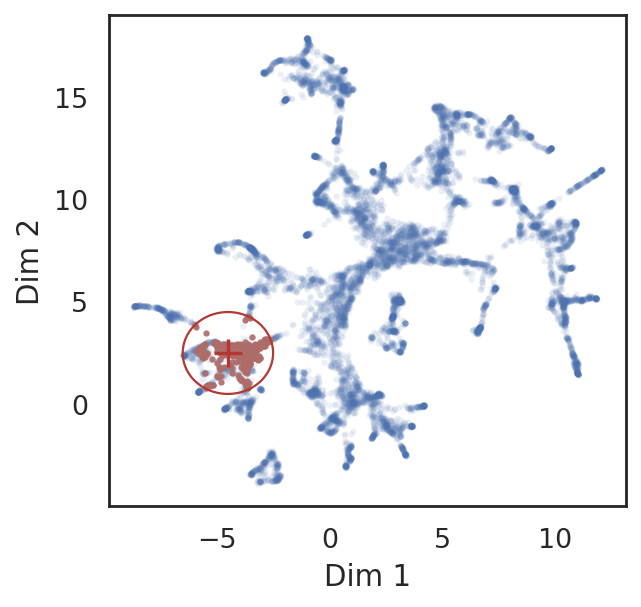
# find neighbor genes of given pins
query_genes = si.tl.query(adata_all_CG,
pin=[-4.5, 2.5],
use_radius=True, r=2,
obsm='X_umap',
anno_filter='entity_anno',
filters=['gene'])
print(query_genes.shape)
query_genes.head()
(293, 5)
[55]:
| celltype | id_dataset | entity_anno | distance | query | |
|---|---|---|---|---|---|
| Lrrc3b | nan | query_1 | gene | 0.112733 | 0 |
| Myct1 | nan | query_1 | gene | 0.143353 | 0 |
| 1810011O10Rik | nan | query_1 | gene | 0.145927 | 0 |
| Tie1 | nan | query_1 | gene | 0.163501 | 0 |
| Sox18 | nan | query_1 | gene | 0.167731 | 0 |
[56]:
# show locations of pin points and its neighbor genes
si.pl.query(adata_all_CG,
show_texts=False,
alpha=0.9,
alpha_bg=0.1,
fig_legend_ncol=1,
fig_size=(4,4))
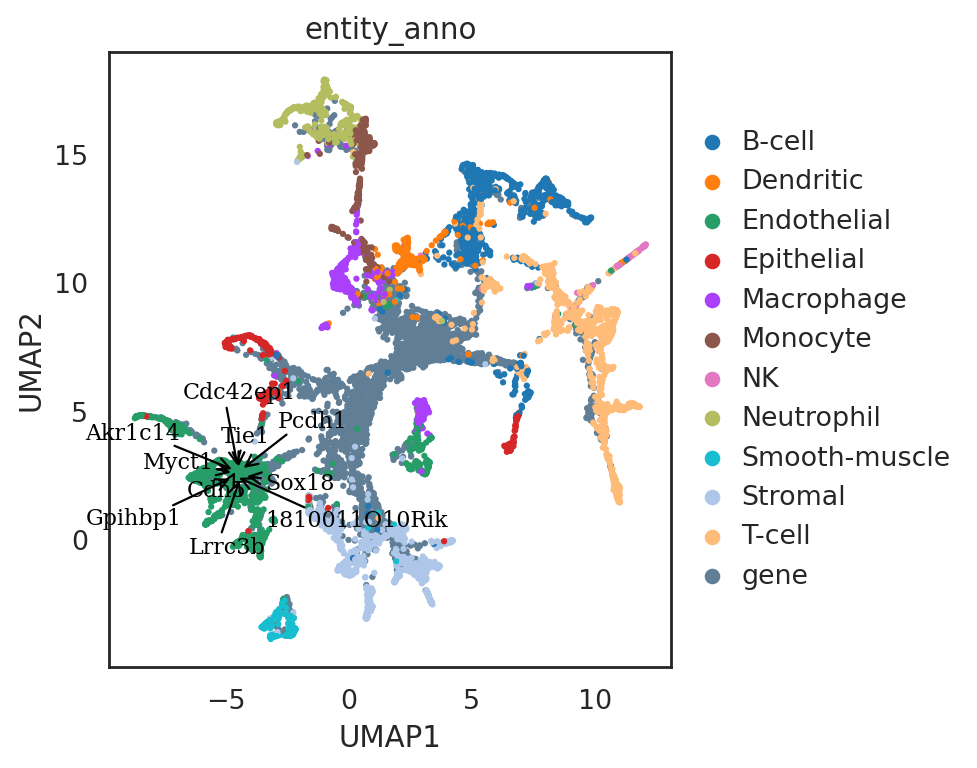
[57]:
si.pl.umap(adata_all_CG[::-1],
color=['entity_anno'],
dict_palette={'entity_anno': palette_entity_anno},
drawing_order='original',
show_texts=True,
texts=query_genes.index[:10],
text_expand=(1.2,1.4),
fig_size=(6,5),
fig_legend_ncol=1)
[ ]:
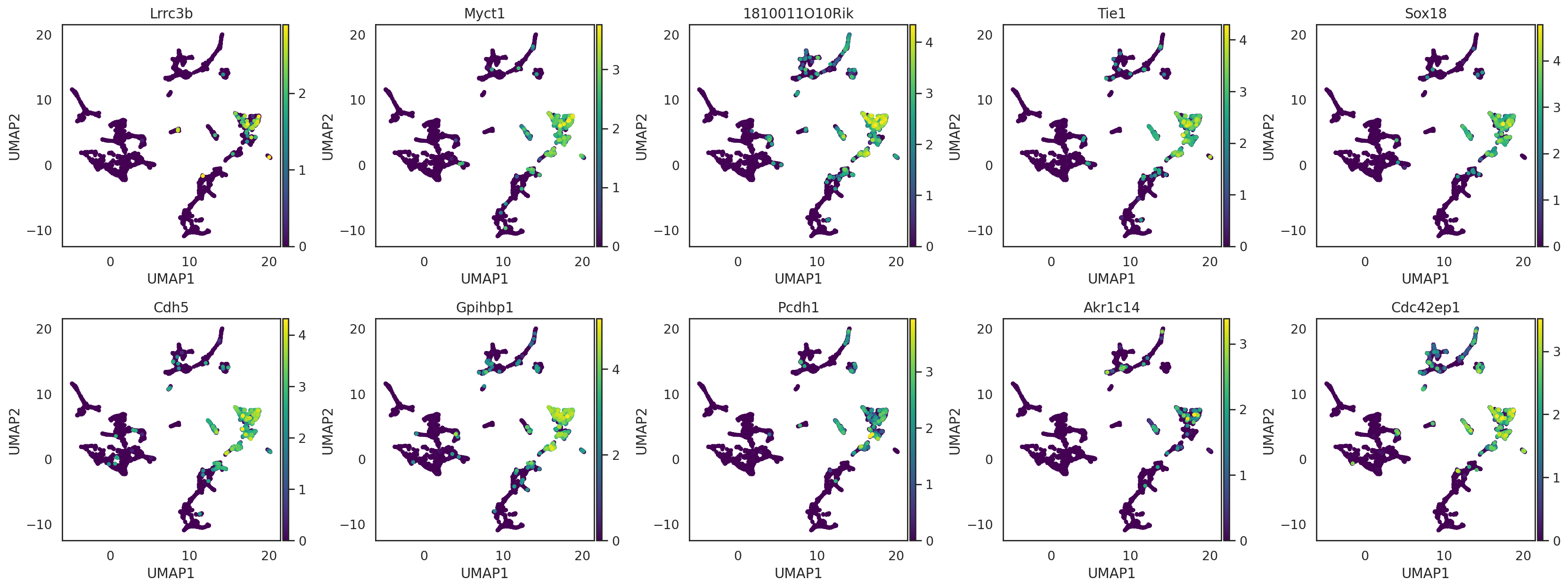
visualize these neighbor genes on UMAP of cells
[58]:
import numpy as np
adata_CG_concat = adata_CG_mi.concatenate([adata_CG_sm],
join='outer',
fill_value=np.nan,
index_unique=None)
adata_CG_concat.obsm['X_umap'] = adata_all[adata_CG_concat.obs_names,].obsm['X_umap'].copy()
/data/pinello/SHARED_SOFTWARE/anaconda_latest/envs/hc_simba/lib/python3.7/site-packages/scipy/sparse/_index.py:125: SparseEfficiencyWarning: Changing the sparsity structure of a csc_matrix is expensive. lil_matrix is more efficient.
self._set_arrayXarray(i, j, x)
/data/pinello/SHARED_SOFTWARE/anaconda_latest/envs/hc_simba/lib/python3.7/site-packages/pandas/core/arrays/categorical.py:2487: FutureWarning: The `inplace` parameter in pandas.Categorical.remove_unused_categories is deprecated and will be removed in a future version.
res = method(*args, **kwargs)
[59]:
si.pl.umap(adata_CG_concat,
color=query_genes.index[:10],
drawing_order='sorted',
fig_ncol=5,
fig_size=(4,4))
[ ]:
save results
[60]:
adata_CG_mi.write(os.path.join(workdir,'adata_CG_mi.h5ad'))
adata_CG_sm.write(os.path.join(workdir,'adata_CG_sm.h5ad'))
adata_C.write(os.path.join(workdir,'adata_C.h5ad'))
adata_C2.write(os.path.join(workdir,'adata_C2.h5ad'))
adata_G.write(os.path.join(workdir,'adata_G.h5ad'))
adata_all.write(os.path.join(workdir,'adata_all.h5ad'))
adata_all_CG.write(os.path.join(workdir,'adata_all_CG.h5ad'))
Read back anndata objects
adata_CG_mi = si.read_h5ad(os.path.join(workdir,'adata_CG_mi.h5ad'))
adata_CG_sm = si.read_h5ad(os.path.join(workdir,'adata_CG_sm.h5ad'))
adata_C = si.read_h5ad(os.path.join(workdir,'adata_C.h5ad'))
adata_C2 = si.read_h5ad(os.path.join(workdir,'adata_C2.h5ad'))
adata_G = si.read_h5ad(os.path.join(workdir,'adata_G.h5ad'))
adata_all = si.read_h5ad(os.path.join(workdir,'adata_all.h5ad'))
adata_all_CG = si.read_h5ad(os.path.join(workdir,'adata_all_CG.h5ad'))
[ ]: